Consultation on common specification requirements for in vitro diagnostic devices
Updated 10 July 2025

© Crown copyright 2025
This publication is licensed under the terms of the Open Government Licence v3.0 except where otherwise stated. To view this licence, visit nationalarchives.gov.uk/doc/open-government-licence/version/3 or write to the Information Policy Team, The National Archives, Kew, London TW9 4DU, or email: psi@nationalarchives.gov.uk.
Where we have identified any third party copyright information you will need to obtain permission from the copyright holders concerned.
This publication is available at https://www.gov.uk/government/consultations/common-specification-requirements-for-in-vitro-diagnostic-devices/consultation-on-common-specification-requirements-for-in-vitro-diagnostic-devices
Ministerial foreword
This consultation is important for Great Britain’s (GB) healthcare system as in vitro diagnostic medical devices (IVD devices) contribute to accurate and timely medical decisions. The IVD industry has advanced at a rapid pace and the Medicines and Healthcare Regulatory products Agency (MHRA) is focused on delivering the future regulatory framework for medical devices. On 26 June 2022, the MHRA published the government response to the public consultation on the future regulation of medical devices in the United Kingdom. Since then, key achievements the MHRA have made include: the establishment of an Innovative Devices Access Pathway pilot, guidance on Software as a medical device, and the laying of regulations for IVD devices in Northern Ireland to lay down proportionate penalties, give the MHRA powers to serve compliance notices for breaches of the IVD Regulations in Northern Ireland, and further strengthens the toolkits for safety of patients.
We are committed to ensuring that our new regime for medical devices prioritises patient safety. We aim to preserve patients’ trust in the GB healthcare system and through this consultation we have an opportunity to ensure that the future regulation of medical devices is centred on patient safety by establishing proportionate regulation.
Executive summary
We want to seek views on the possible amendments to the Medical Devices Regulations 2002 to include common specification requirements for manufacturers of high-risk IVD devices. This consultation builds upon public views after an initial consultation in November 2021 and will help inform further policy decision-making in this area. The scope of this consultation is specific to the common specification requirements for high-risk IVD devices. The proposals provide additional detail of the essential requirements for applicable high-risk IVD devices in addition to those that were consulted on in November 2021. Through the response to the consultation, we outlined intentions to be consistent with the General Safety and Performance Requirements (GSPR) in the In Vitro Diagnostic Medical Device Regulation 2017/746 (IVDR) which was updated in 2022 to include common specification requirements for certain high-risk IVD devices. The aim of the public consultation is to explore whether there is support for the inclusion of common specification requirements and, if there is, appetite to be consistent with the IVDR requirements.
The common specification requirements include requirements for COVID-19 test devices, which are scientifically more stringent than current Great Britain requirements that were added into the Medical Devices Regulations 2002 by the Medical Devices (Coronavirus Test Device Approvals) (Amendment) Regulations 2021. Therefore, we are also seeking views in this consultation on the removal of current Coronavirus Test Device Approvals (CTDA) requirements. This will avoid regulatory duplication and unnecessary burden on IVD businesses, as they would not need to go through the CTDA process as well as meeting the common specification requirements.
We will also continue to engage with key stakeholders in the IVD industry and with patients to ensure there is wide representation and transparency in our policy decision-making. We believe that it is critical to ensure that patients have access to IVD devices that are safe, and our regulation is risk proportionate, beneficial, and of a high standard.
Introduction
The in vitro diagnostic medical device (IVD device) industry is a vital part of the UK’s healthcare system. IVD devices play a key role by contributing to accurate and timely medical decisions. An IVD device is used to examine samples taken from the human body. Samples can include saliva, blood, tissue, urine. They are used to diagnose and monitor health conditions such as flu infections, glucose levels, and hepatitis. They can also be used as a prevention measure by screening people for possible health conditions such as cancer. Examples of IVD devices include pregnancy tests, pap smears tests for cervical cancer screening, blood glucose meters for monitoring diabetes, and HIV testing kits.
IVD devices in GB (England, Wales, and Scotland) are currently regulated by the Medicines and Medical Devices Act 2021 and the Medical Devices Regulations 2002. Under the terms of the Windsor Framework, Northern Ireland continues to follow EU regulations for IVD devices, the IVDR. On 21 March 2024, The Medical Devices In Vitro Diagnostic Devices etc. Amendment Regulations 2024 came into force and introduced provisions required for implementing the IVDR in Northern Ireland.
Under the Medical Devices Regulations 2002, an IVD device cannot be placed on the market or put into service in GB without having an affixed UKCA marking (with some exceptions - e.g. custom-made devices and CE marked devices). Manufacturers of IVD devices must ensure their products meet relevant regulatory requirements before placing them on the market or putting them into service. Medium to high risk IVD devices (class B, C and D devices under the future IVD classification system) must undergo conformity assessment from a UK Approved Body to demonstrate conformity to the relevant regulatory requirements for the IVD device, before a UKCA marking can be affixed. This requires manufacturers to demonstrate conformity to the required standards and to also provide information to support their performance claims for their IVD device. In many cases, this information will come from a performance study of the device.
Once a medical device is on the market, the manufacturer must continue to assess the safety and performance of that IVD device. This is known as ‘post-market surveillance’. The manufacturer should also report certain incidents involving the IVD device to the MHRA. This is known as ‘vigilance’. Read more about existing requirements for placing a IVD device on the UK market.
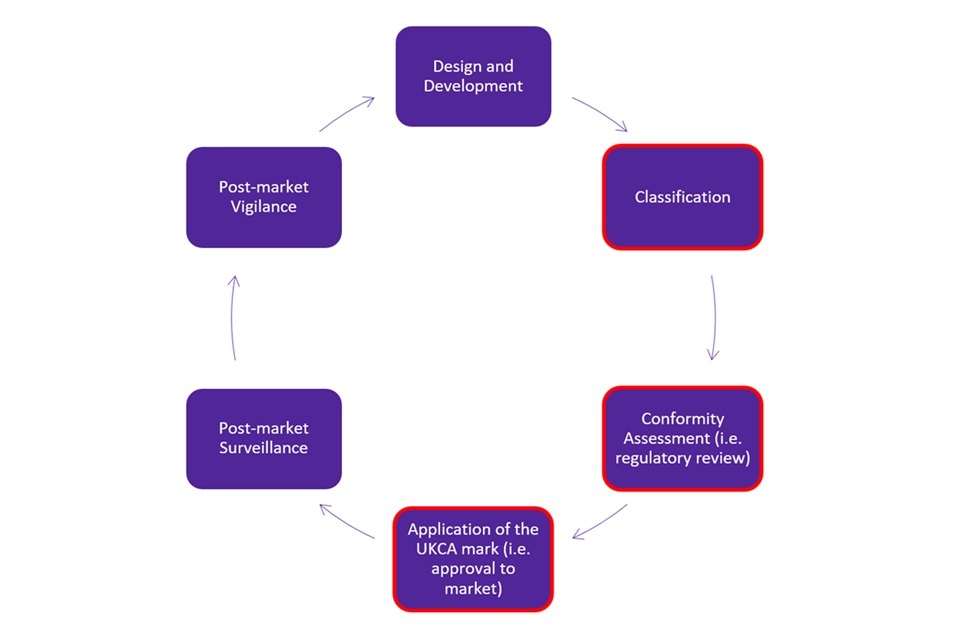
The lifecycle of an IVD device can be found in Figure 1. This consultation does not cover the regulation of the full lifecycle of an IVD device. It covers pre-market approval of IVD devices (boxes outlined in red in Figure 1). In particular, it covers the technical requirements for certain high-risk IVD devices that manufacturers must demonstrate conformity with before being able to receive a UKCA marking.
Figure 1. Life cycle of an in vitro diagnostic medical device
Policy objectives
We want to develop a future regime for IVD devices which enables:
- Improved patient and public safety
- Greater transparency of regulatory decision making and medical device information
- Close alignment with international best practice, and
- More proportionate regulation of medical devices.
The MHRA is inviting members of the public, including the views of patients, medical device researchers, developers, manufacturers and suppliers, clinicians, other healthcare professionals to provide their views on the introduction of common specification requirements to the regulatory framework for IVD devices in GB which will also replace the COVID-19 test approval process.
Common specifications
The Medicines and Medical Devices Act 2021 allows for amendments to the regulations for in vitro diagnostic (IVD) devices. We are seeking views to include common specification requirements in the Medical Devices Regulations 2002 (MDR 2002) before certain high risk IVD devices can receive a UKCA marking and be placed on the GB market, and to remove requirements for COVID-19 test devices from the MDR 2002.
Common specification requirements are a minimum set of performance expectations. The purpose of common specification requirements is to ensure that where there are public health concerns on the safety or performance of certain high-risk IVD devices, these can be addressed by requiring manufacturers to demonstrate that their device complies with the common specification requirements. These are highly specific technical requirements for certain high-risk IVD devices[footnote 1].
Common specifications is not a new policy initiative.
‘Common specifications’ is a similar concept to ‘common technical specifications’, which are already in place for certain high-risk IVD devices under the current MDR 2002. Common technical specifications are minimum performance requirements that must be met as essential requirements before placing certain high-risk IVD devices on the GB market (i.e. it is the regulatory requirements a manufacturer must demonstrate conformity to before being able to receive a UKCA marking). This requirement applies to certain high risk IVD devices (List A devices Annex II), including tests to detect HIV, hepatitis, blood grouping and human T-Cell lymphotropic virus. An IVD device must undergo a conformity assessment by an approved body, which is dependent on meeting the common technical specifications. The common technical specifications can also be used for performance evaluations and re-evaluation.
The Medical Device Regulations are undergoing classification amendments.
The future regulatory amendments to the MDR 2002 will update the IVD classification system using a risk-based approach, with Class A being lowest risk class devices and Class D being highest risk class devices. This classification system adopts the principles of harmonisation to internationally-recognised principles agreed by the International Medical Device Regulators Forum (IMDRF) and classification system used in the EU. Therefore, the common technical specifications referenced in the current MDR 2002 applying to List A devices will become obsolete.
We propose the introduction of common specification requirements as a means for certain class D IVD devices in the future regulation to demonstrate conformity with applicable essential requirements, in the same way that the common technical specifications apply to Annex II List A IVD devices under current regulations. Common specification requirements build upon common technical specifications, reflecting the latest scientific and technical progress with updated general and specific requirements related to performance characteristics for each disease and the concept of rapid tests. Specifications for SARS-CoV-2, Creutzfeldt-Jakob, cytomegalovirus, Barr-Epstein, syphilis and trypanosoma cruzi diseases have also been added.
Figure 2. Comparison of the current and future UK alignment
| Current UK MDR 2002 | Future UK MDR 2025 | |
|---|---|---|
| 1. Classification system: predetermined list (e.g. List A high risk IVDs) | → | 1. Classification system: Risked-based classification (e.g. Class D high risk IVDs) |
| 2. Common technical specifications for certain List A IVDs | → | 2. Common specifications for certain Class D IVDs |
The inclusion of common specification requirements in the MDR 2002 removes the need for COVID-19 test device requirements.
Common specification requirements will include quality and technical requirements for Coronavirus (SARS-CoV-2) tests. It would be duplicative with the Coronavirus Test Devices Approvals (CTDA) regulations in the current MDR 2002. In July 2021, the government introduced requirements in the MDR 2002 to govern the validation of COVID-19 tests in response to variation in test performance consistency and quality. The aim of the CTDA policy was to ensure that COVID-19 tests adhered to minimum quality and technical standards so there were consistently high-quality tests made available at the height of the pandemic. The policy was to ensure that all mature antigen and molecular detection COVID-19 test devices adhere to minimum standards of performance as set out in regulation 38B of the MDR 2002, to reduce the potential risk to public health. The CTDA process currently requires antigen and molecular COVID-19 test devices to undergo mandatory desktop review by the MHRA.
In December 2022, the government conducted a review of the CTDA process to establish the impact of the legislation, identify any lessons learnt, and discern whether the policy had met its key objectives. The review found that the policy was successful in meeting its principal and overall objective of improving the quality of tests on the UK market and addressed the market failure that saw poor quality, inaccurate tests made available for sale.
The common specification requirements are an equivalent or better specification that also meets the CTDA policy objectives. Common specification requirements for Coronavirus would eliminate the need for COVID-19 tests to go through the CTDA process and will require approved bodies as part of the conformity assessment process to assess whether the COVID-19 test devices meets the common specification requirements. Therefore, IVD businesses would only need to meet common specifications for Coronavirus requirements, avoiding regulatory duplication and unnecessary burdens to manufacturers.
Common specification requirements policy proposal for Great Britain
As mentioned above, the classification system for IVD devices in GB will be changing from a predetermined list to a risk-based classification system. This means that the common technical specifications for high-risk List A IVD devices will no longer be applicable as the devices will be reclassified as Class D IVD devices. This leaves a gap in regulatory requirements for high-risk IVD devices and poses a risk on patient safety.
Common technical specifications ensure that technical requirements are highly specific for high-risk IVD devices and provide a way for manufacturers help ensure that their devices are safe for patients. Therefore, we believe that it is important to continue to have these safety requirements in place and protect patients.
In the November 2021 consultation we received support for amending the essential requirements of the MDR 2002, with a desire to be consistent with the General Safety and Performance Requirements (GSPR) in the IVDR. As set out in Article 9 and Annex II of the EU IVDR, the use of harmonised standards and common specifications are required for demonstrating conformity with GSPRs. In 2022, the EU introduced a new piece of regulation, to include common specification requirements for certain high-risk IVD devices. Therefore, listening to the support to be consistent with the GSPRs, we are proposing to adopt common specification requirements.
We propose:
-
To introduce common specification requirements for certain Class D IVD devices to be consistent with EU Commission Implementing Regulation 2022/1107. This is to ensure that manufacturers have enough safety checks in place for certain Class D devices that are the highest risk to patient safety. Proper application of common specification ensures that certain high-risk IVD devices meet a common benchmark requirement. This is to ensure a continuous high level of safety and performance of high risk IVD devices. It would also align GB IVD requirements with Northern Ireland. In practice, it would mean that during conformity assessment of certain high-risk IVD devices, Approved Bodies will be required to assess the performance of the IVD in meeting the common specification requirements where no harmonised standard exists.
-
To introduce the inclusion of common specification requirements in a Post Market Performance Follow-up (PMPF) Plan. In the IVDR, a PMPF plan must be submitted as part of the technical requirements for an IVD device before it can be placed on the market. It is crucial to have this plan in place to ensure that there is ongoing monitoring of the safety and effectiveness of IVD devices throughout their lifecycle, once they have been placed on GB market. In practice, it would mean that during conformity assessment of high risk IVD devices, approved bodies will be required to assess the PMPF plan and ensure it includes monitoring for common specifications. We previously consulted on PMPF requirements for IVD devices in the November 2021 consultation.
-
To remove the requirements for COVID-19 test devices from the MDR 2002. This will avoid regulatory duplication and unnecessary burden on IVD businesses and further align with the regulatory framework in Northern Ireland. In practice, this would mean that an IVD manufacturer planning to place a COVID-19 test device on the Great Britain market would only need to comply with common specifications for SARS-CoV-2 tests and not the Coronavirus Test Device Approvals requirements in the MDR 2002. Approved Bodies would be required to assess the performance of the COVID-19 test device in meeting the common specification requirements, rather than the Coronavirus Test Device Approvals assessment currently being managed by MHRA.
Consultation questions
C1. The government proposes that the Medical Devices Regulations 2002 incorporates the common specifications for certain Class D IVDs, as set out in the Commission Implementing Regulation (EU) 2022/1107. Do you agree with this proposal?
- Yes
- No, please provide any further comments you may have (short text)
- No opinion
C2. Do you think meeting common specifications requirements should be a requirement in a Post Market Performance Follow-up Plan?
- Yes
- No, please provide any further comments you may have (short text)
- No opinion
C3. The government proposes to remove the requirements for Coronavirus test devices, from the MDR 2002. The government proposes to require COVID-19 test devices to undergo a conformity assessment by an approved body, meeting common specifications requirements in line with Commission Implementing Regulation (EU) 2022/1107, the EU common specifications for certain Class D IVDs. Do you agree with this proposal?
- Yes
- No, please provide any further comments you may have (short text)
- No opinion
Demographic questions - about you
D1A. In which capacity are you primarily responding to this survey?
a. An individual sharing my personal views and experiences
b. An individual sharing my professional views
c. On behalf of an organisation
[if selected D1A.a. or D1A.b.] D1B. Are you currently working as a clinical professional? Which of the below describes you best?
a. No, I’m not a clinical professional
b. Yes, I’m a [free text box with 50 character limit]
c. Other (please specify)
[if selected D1A.b. or D1A.c.] D1C. Which of the below describes your organisation best?
a. Trade association
b. Business
c. Patient group
d. Professional representative group
e. Professional regulator
f. Research organisation
g. Other (please specify)
[if selected D1A.a.] D2A. Where do you live in the UK?
a. England
b. Northern Ireland
c. Scotland
d. Wales
e. I live outside the UK
[if selected D1A.b. or D1A.c.] D2B. Where does your organisation operate? (Please tick all that apply)
a. England
b. Scotland
c. Wales
d. Northern Ireland
e. Outside the UK
[if selected D1A.b. or D1A.c.] D2D. How many employees does your business employ? An employee is anyone aged 16 years or over that an organisation directly pays from its payroll(s), in return for carrying out a full-time or part-time job or being on a training scheme. It excludes voluntary workers, self-employed and working owners who are not paid via PAYE.
a. 0-9
b. 10-49
c. 50-249
d. 250-499
e. 500+
f. Don’t know
[if selected D1A.b., D1A.c., or D1A.g.] D2E. Does your business produce or supply any of the following products?
a. Medicines
b. Medical devices
c. In vitro diagnostic medical devices
d. Borderline substances (e.g. medical nutrition)
e. None of the above [exclusive]
f. Don’t know [exclusive]
[if selected D2E.c.] D2F. Does your business produce or supply any of the devices affected by common specification measures?
a. COVID-19 tests
b. Other
c. Don’t know
Satisfaction survey - give feedback on participating
Instruction text - If you do not wish to leave your feedback, please select the ‘Submit’ button.
S1. It was easy to participate in this opportunity
a. Strongly agree
b. Agree
c. Neither agree or disagree
d. Disagree
e. Strongly disagree
S2. The supporting information was understandable
a. Strongly agree
b. Agree
c. Neither agree or disagree
d. Disagree
e. Strongly disagree
S3. What could we do better?
- (Free text box)
Data protection notice
Over the month of May 2024, the Medicines and Healthcare products Regulatory Agency will seek the views of individuals and organisations through a public consultation, to inform amendments to the Medical Devices Regulations 2002 to include common specifications requirements for high risk in vitro diagnostic medical devices.
This notice sets out how data collected through this call for evidence will be used and respondents’ rights under Articles 13 and/or 14 the UK General Data Protection Regulation (GDPR).
Data controller
The Medicines and Healthcare products Regulatory Agency (MHRA) is the data controller.
What personal data we collect
You can respond to the consultation through our public survey, which can be completed online. Alternatively, you can download the form, complete it and send this to us by email.
We will collect data on:
- whether you are responding as an individual or on behalf of an organisation.
- your occupation
- your name and name of your organisation
- the country and region you live in, or where your organisation provides services in the UK
- your ethnic group, if provided
If volunteered by you, we will also collect data on:
- your email address (if completing a paper survey and submitting it by email, or if responding on behalf of an organisation and confirming MHRA can contact you about your response)
- any other personal data you volunteer by way of evidence or example in your response to open-ended questions in the survey
How we use your data (purposes)
Your data will be treated in the strictest of confidence.
We collect your personal data as part of the consultation process:
- for statistical purposes, for example, to understand how representative the results are and whether views and experiences vary across demographics
- so that MHRA can contact you for further information about your response (if you are responding on behalf of an organisation and have given your consent)
Legal basis for processing personal data
The legal basis for processing your personal data is to perform a task carried out in the public interest, or in the exercise of official authority vested in the controller.
Data processors and other recipients of personal data
All responses to the consultation will be seen by:
- professionals in MHRA who are working on this consultation
- MHRA’s third-party supplier (SocialOptic), who is responsible for running and hosting the online survey
No personally identifiable data will be shared.
MHRA may also share your responses, when anonymised, with Department of Health and Social Care, Government Legal Department, Office for Life Sciences, and any other government body identified to be part of this consultation
International data transfers and storage locations
Storage of data by MHRA is provided via secure computing infrastructure on servers located in the UK. Our platforms are subject to extensive security protections and encryption measures.
Storage of data by SurveyOptic is provided via secure servers located in the United Kingdom (UK).
Retention and disposal policy
Personal data will be held by MHRA for 3 years and disposed of sooner if possible.
SurveyOptic will securely erase the data held on their system 5 years after the call for evidence online survey closes, or when instructed to do so by MHRA if the data has served its intended purpose (whichever happens earlier).
Data retention will be reviewed on an annual basis. Anonymised data may be kept indefinitely.
How we keep your data secure
MHRA use appropriate technical, organisational and administrative security measures to protect any information we hold in our records from loss, misuse, unauthorised access, disclosure, alteration and destruction. We have written procedures and policies which are regularly audited and reviewed at a senior level.
SurveyOptic is Cyber Essentials certified.
Your rights as a data subject
By law, you have rights as a data subject. Your rights under the UK General Data Protection Regulation and the UK Data Protection Act 2018 apply.
These rights are:
- the right to get copies of information – individuals have the right to ask for a copy of any information about them that is used
- the right to get information corrected – individuals have the right to ask for any information held about them that they think is inaccurate, to be corrected
- the right to limit how the information is used – individuals have the right to ask for any of the information held about them to be restricted, for example, if they think inaccurate information is being used
- the right to object to the information being used – individuals can ask for any information held about them to not be used. However, this is not an absolute right, and continued use of the information may be necessary, with individuals being advised if this is the case
- the right to get information deleted – this is not an absolute right, and continued use of the information may be necessary, with individuals being advised if this is the case
Comments or complaints
Anyone unhappy or wishing to complain about how personal data is used as part of this programme, should contact dataprotection@mhra.gov.uk in the first instance or write to:
Data Protection Officer
MHRA
10 South Colonnade
London E14 4PU
Anyone who is still not satisfied can complain to the Information Commissioner’s Office. Their website address is www.ico.org.uk and their postal address is:
Information Commissioner’s Office
Wycliffe House
Water Lane
Wilmslow
Cheshire
SK9 5AF
© Crown copyright 2024
Produced by the Medicines and Healthcare products Regulatory Agency. www.gov.uk/mhra
This publication is licensed under the terms of the Open Government Licence. To view this licence, visit http://www.nationalarchives.gov.uk/doc/open-government-licence or email: psi@nationalarchives.gov.uk.
The names, images and logos identifying the Medicines and Healthcare products Regulatory Agency are proprietary marks. All the Agency’s logos are registered trademarks and cannot be used without the Agency’s explicit permission.
Annex A: Legal definitions
Medical devices are defined in regulation 2(1) of the UK Medical Devices Regulations 2002 (as amended), as follows:
“medical device” means an instrument, apparatus, appliance, material or other article, whether used alone or in combination, together with any software necessary for its proper application, which—
(a) is intended by the manufacturer to be used for human beings for the purpose of-
(i) diagnosis, prevention, monitoring, treatment or alleviation of disease,
(ii) diagnosis, monitoring, treatment, alleviation of or compensation for an injury or handicap,
(iii) investigation, replacement or modification of the anatomy or of a physiological process, or
(iv) control of conception; and
(b) does not achieve its principal intended action in or on the human body by pharmacological, immunological or metabolic means, even if it is assisted in its function by such means,
and includes devices intended to administer a medicinal product or which incorporate as an integral part a substance which, if used separately, would be a medicinal product and which is liable to act upon the body with action ancillary to that of the device;
IVDs are defined in regulation 2(1) of the UK Medical Devices Regulations 2002 (as ammeded), as follows:
“in vitro diagnostic medical device” means a medical device which—
(a). is a reagent, reagent product, calibrator, control material, kit, instrument, apparatus, equipment or system, whether used alone or in combination; and
(b) is intended by the manufacturer to be used in vitro for the examination of specimens, including blood and tissue donations, derived from the human body, solely or principally for the purpose of providing information—
(i) concerning a physiological or pathological state,
(ii) concerning a congenital abnormality,
(iii) to determine the safety and compatibility of donations, including blood and tissue donations, with potential recipients, or
(iv) to monitor therapeutic measures,
and includes a specimen receptacle but not a product for general laboratory use, unless that product, in view of its characteristics, is specifically intended by its manufacturer to be used for in vitro diagnostic examination.
Annex B: Legal basis and the assessment of the matters set out in section 15 of the Medicines and Medical Devices Act 2021
The Medicines and Medical Devices Act 2021 (‘the Act’) received Royal Assent on 11 February 2021. We propose to make the legislative changes under consultation in this document, under Part 4 of the Act, which provides powers to make regulations about medical devices.
This consultation is conducted in line with the consultation requirements in section 15 of the Act. Section 15 of the Act states that in making regulations the overarching objective must be safeguarding public health and requires when considering whether regulations contribute to this objective, the Secretary of State must have regard to:
- the safety of medical devices;
- the availability of medical devices;
- the likelihood of the United Kingdom being seen as a favourable place in which to:
a. carry out research relating to medical devices,
b. develop medical devices, or
c. manufacture or supply medical devices.
We have assessed the proposal against each of these factors, outlined below.
We are proposing to introduce Common Specifications for certain high-risk IVD devices (including COVID-19 tests) by being consistent with the IVDR and to remove the CTDA process for COVID-19 devices.
Safety: The purpose of the Common Specifications policy is to improve the safety profile of certain high risk IVD devices (including Coronavirus Test Devices) to protect patient safety. This policy would require manufacturers to demonstrate that their device complies with the Common Specifications where there are public health concerns on the safety or performance of a device. Common Specifications will also include scientifically more stringent safety requirements for COVID-19 tests than CTDA.
Availability: Common Specifications will remove the need for COVID-19 detection tests to go through an additional market access route (the CTDA process) in comparison to other high-risk IVD devices. COVID-19 test device manufacturers would only need to meet Common Specifications requirements, avoiding regulatory duplication and unnecessary burden on IVD businesses.
Favourability: The November 2021 consultation revealed strong support to be consistent with the General Safety and Performance Requirements (GSPR) in the IVDR, and this proposal ensures that specific technical requirements are imposed whilst maintaining consistency with the approach taken in the EU. Consistency with EU regulations in this area would help support the availability and favourability of the Great Britain market.
-
The list of high-risk IVD devices includes devices detecting and/or quantifying HIV, human T-cell lymphotropic virus, hepatitis B, C, D, variant Creutzfeldt-Jakob disease, cytomegalovirus, Epstein-Barr virus, Treponema pallidum (causative agent of syphilis), Trypanosoma cruzi (causative agent of Chagas disease) and SARS-CoV-2, as well as for determining ABO, Rhesus, Kell, Kidd and Duffy blood groups. ↩