Abrocitinib: Treatment protocol: Information for healthcare professionals
Published 2 February 2021

© Crown copyright 2021
This publication is licensed under the terms of the Open Government Licence v3.0 except where otherwise stated. To view this licence, visit nationalarchives.gov.uk/doc/open-government-licence/version/3 or write to the Information Policy Team, The National Archives, Kew, London TW9 4DU, or email: psi@nationalarchives.gov.uk.
Where we have identified any third party copyright information you will need to obtain permission from the copyright holders concerned.
This publication is available at https://www.gov.uk/government/publications/abrocitinib-in-the-treatment-of-severe-atopic-dermatitis/abrocitinib-treatment-protocol-information-for-healthcare-professionals
Early Access to Medicines Scheme – Treatment protocol – Information for healthcare professionals
Introduction
The aim of the Early Access to Medicines Scheme (EAMS) is to provide earlier availability of promising new unlicensed medicines and medicines used outside their licence, to UK patients that have a high unmet clinical need. The medicinal products included in the scheme are those that are intended to treat, diagnose or prevent seriously debilitating or life-threatening conditions where there are no adequate treatment options. More information about the scheme.
This information is intended for healthcare professionals and is provided by the pharmaceutical company that manufactures the EAMS medicine. This medicine does not yet have a licence (marketing authorisation) and the information is provided to assist physicians in prescribing this unlicensed medicine. Guidance on prescribing unlicensed medicines can be found on the GMC webpage.
The scientific opinion is based on assessment of the information supplied to the MHRA on the benefits and risks of this promising new medicine. As such, this is a scientific opinion and should not be regarded as a medicine licensed by the MHRA or a future commitment by the MHRA to license such a medicine, nor should it be regarded as an authorisation to sell or supply such a medicine. A positive scientific opinion is not a recommendation for use of the medicine and should not be interpreted as such. Under EAMS the risk and legal responsibility for prescribing a ‘special’ remains with the physician, and the opinion and EAMS documentation published by the MHRA are intended only to inform physicians’ decision making and not to recommend use. An EAMS scientific opinion does not affect the civil liability of the manufacturer or any physician in relation to the product.
Healthcare professionals should also refer to the summary information on the pharmacovigilance system which is provided in the document ‘Early Access to Medicines Scheme – Treatment protocol – Information on the pharmacovigilance system’.
Scientific opinion period: The MHRA will withdraw the EAMS positive scientific opinion when a marketing authorisation (drug licence) is issued for the EAMS product covering the EAMS indication, or if following scientific assessment, the EAMS criteria are considered to be no longer met.
Treatment protocol update(s): In case of substantial new efficacy or safety data, the treatment protocol may need to be updated.
Contact information regarding queries on using this EAMS medicine can be found at the end of this document.
Information for healthcare professionals
1. Name of the medicinal product
Abrocitinib 100 mg film-coated tablets
2. Qualitative and quantitative composition
Abrocitinib 100 mg film-coated tablets:
Each film-coated tablet contains 100 mg abrocitinib.
Excipient with known effect:
Each abrocitinib 100 mg film-coated tablet contains 2.73 mg of lactose monohydrate, corresponding to the amount of lactose monohydrate in the film coating material Opadry® II White 33G28523.
For the full list of excipients, see section 6.1.
3. Pharmaceutical form
Abrocitinib 100 mg film-coated tablets (tablet):
White, round, film-coated tablet.
4. Clinical particulars
4.1 EAMS therapeutic indication
Abrocitinib is indicated for the treatment of adult and adolescent patients with severe atopic dermatitis requiring treatment with systemic therapy and have had inadequate response or have lost response to approved systemic therapies, or those who are ineligible or intolerant of approved systemic therapies.
4.2 Posology and method of administration
Treatment should be initiated and supervised by a healthcare professional experienced in the diagnosis and treatment of atopic dermatitis.
Posology
For most patients who have had an inadequate response to available systemic therapies, the starting dose should be 200 mg. For patients who have risk factors for developing an adverse reaction to abrocitinib (e.g. age over 65 years) or who are less likely to tolerate the adverse reactions, a starting dose of 100 mg should be considered (see section 4.4 and 4.8).
Abrocitinib can be used with or without medicated topical therapies for atopic dermatitis.
Discontinuation of abrocitinib treatment should be considered if there is no evidence of therapeutic benefit after 12 weeks of treatment.
Treatment with abrocitinib should not be initiated in patients with a platelet count <150 × 103/mm3, an absolute lymphocyte count (ALC) <0.5 × 103/mm3, an absolute neutrophil count (ANC) <1 × 103/mm3, or who have a haemoglobin value less than 8 g/dL (see section 4.4).
Missed doses
If a dose is missed, patients should be advised to take the dose as soon as possible unless it is less than 12 hours before the next dose, in which case the patient should not take the missed dose. Thereafter, resume dosing at the regular scheduled time.
Dose interruption
If a patient develops a serious infection, sepsis or opportunistic infection, interruption of abrocitinib until the infection is controlled should be considered (see section 4.4).
Interruption of dosing may be needed for management of laboratory abnormalities as described in Table 2 (see section 4.4).
Drug-drug interactions
When indicated dose is 200 mg (2 x 100 mg tablets) abrocitinib once daily, the dose should be reduced by 50% to 100 mg (1 x 100 mg tablet) once daily in patients receiving strong inhibitors of cytochrome P450 (CYP) 2C19 (e.g. fluvoxamine, fluconazole, fluoxetine). The use of abrocitinib is not recommended concomitantly with strong inducers of CYP enzymes (e.g. rifampin) (See section 4.5).
When indicated abrocitinib dose is 100 mg, patients receiving any of the above-mentioned CYP inhibitors will not be able to initiate treatment under the EAMS as the 100 mg tablet is the lowest strength available as part of the EAMS.
Special populations
Renal impairment No dose adjustment is required in patients with mild renal impairment, i.e. estimated glomerular filtration rate (eGFR) of 60 to <90 mL/min. In patients with moderate (eGFR 30 to <60 mL/min) or severe (eGFR <30 mL/min) renal impairment, the recommended dose of abrocitinib is reduced by 50% as shown in Table 1 (see section 5.2). When the required dose is 100mg (i.e. as selected for patients with normal renal function) patients with moderate or severe renal impairment will not be able to initiate treatment under EAMS as the 100 mg tablet is the lowest strength available as part of the EAMS. The use of abrocitinib has not been studied in patients with end-stage renal disease (ESRD) on renal replacement therapy.
Table 1. Dose adjustments for renal impairment
| Renal impairment stage | Estimated glomerular filtration rate (eGFR) | Indicated dose 100 mg once daily | Indicated dose 200 mg once daily |
|---|---|---|---|
| Mild | 60 to <90 mL/min | None | None |
| Moderate | 30 to <60 mL/min | Not able to initiate treatment as part of the EAMS | Abrocitinib 100 mg once daily |
| Severe | <30 mL/min | Not able to initiate treatment as part of the EAMS | Abrocitinib 100 mg once daily |
Hepatic impairment No dose adjustment is required in patients with mild (Child-Pugh A) or moderate (Child-Pugh B) hepatic impairment. Abrocitinib has not been studied in patients with severe (Child-Pugh C) hepatic impairment and is therefore not recommended (see section 5.2).
Elderly No dose adjustment is recommended for patients ≥65 years of age. There are limited data in patients 75 years of age and older.
Paediatric population The safety and efficacy of abrocitinib in paediatric patients under 12 years of age have not yet been established. No data are available. Patients under 12 years of age will not be able to initiate treatment under the EAMS.
Method of administration
Abrocitinib is to be taken orally once daily (one tablet for 100 mg, or two tablets for 200 mg at the same time) with or without food at approximately the same time each day.
In patients who experience nausea while taking abrocitinib, taking with food may improve nausea.
Abrocitinib tablets should be swallowed whole with water and should not be split, crushed, or chewed.
4.3 Contraindications
Hypersensitivity to the active substance or to any of the excipients listed in section 6.1. Pregnancy (see section 4.6).
4.4 Special warnings and precautions for use
Serious infections
Serious infections have been reported in patients receiving abrocitinib. The most frequent serious infections in clinical studies were herpes simplex, herpes zoster, and pneumonia, (see section 4.8). The risks and benefits of treatment with abrocitinib should be carefully considered prior to initiating in patients with active, chronic or recurrent infections.
Patients should be closely monitored for the development of signs and symptoms of infection during and after treatment with abrocitinib. A patient who develops a new infection during treatment with abrocitinib should undergo prompt and complete diagnostic testing and appropriate antimicrobial therapy should be initiated. The patient should be closely monitored, and abrocitinib therapy should be interrupted if the patient is not responding to standard therapy.
Tuberculosis Patients should be screened for tuberculosis (TB) before starting abrocitinib therapy and consider yearly screening for patients in highly endemic areas for TB. Abrocitinib should not be given to patients with active TB. For patients with a new diagnosis of latent TB or prior untreated latent TB, preventive therapy for latent TB should be started prior to initiation of abrocitinib.
Viral reactivation Viral reactivation, including herpes virus reactivation (e.g., herpes zoster, herpes simplex), was reported in clinical studies (see section 4.8). The rate of herpes zoster infections was higher in patients 65 years of age and older (see section 4.8).
Eczema herpeticum (disseminated viral infection mostly due to herpes simplex virus) was also reported in clinical studies with abrocitinib. The condition is characterised by fever, malaise and rapid spread of vesicular and erosive lesions in patients with atopic dermatitis and requires prompt treatment with antiviral agents. Discontinuation or interruption of abrocitinib therapy until the resolution of an eczema herpeticum infection should be considered, depending on the seriousness of the event.
Screening for viral hepatitis should be performed in accordance with clinical guidelines before starting therapy and during therapy with abrocitinib. Patients with evidence of active hepatitis B or hepatitis C (positive hepatitis C PCR) infection were excluded from clinical studies (see section 5.2). Patients who were hepatitis B surface antigen negative, hepatitis B core antibody positive, and hepatitis B surface antibody positive had testing for hepatitis B virus (HBV) DNA. Patients who had HBV DNA above the lower limit of quantification (LLQ) were excluded. Patients who had HBV DNA negative or below LLQ could initiate treatment with abrocitinib; such patients had HBV DNA monitored. If HBV DNA is detected, a liver specialist should be consulted to determine if treatment interruption is warranted.
Vaccination
No data are available on the response to vaccination with live vaccines in patients receiving abrocitinib. The use of live, attenuated vaccines during or immediately prior to abrocitinib therapy is not recommended. Prior to initiating abrocitinib it is recommended that patients be brought up to date with all immunisations, including prophylactic herpes zoster vaccinations, in agreement with current immunisation guidelines.
Venous thromboembolism
Events of deep venous thrombosis (DVT) and pulmonary embolism (PE) have been reported in patients receiving Janus kinase (JAK) inhibitors including abrocitinib (see section 4.8). Abrocitinib should be used with caution in patients at high risk for DVT/PE. Risk factors that should be considered in determining the patient’s risk for DVT/PE include older age, obesity, a medical history of DVT/PE, prothrombotic disorder, use of combined hormonal contraceptives or hormone replacement therapy, patients undergoing major surgery, or prolonged immobilisation. If clinical features of DVT/PE occur, abrocitinib treatment should be discontinued and patients should be evaluated promptly, followed by appropriate treatment.
Malignancy (including non-melanoma skin cancers)
Malignancies, including non-melanoma skin cancer (NMSC), were observed in clinical studies with abrocitinib. Clinical data are insufficient to assess the potential relationship of exposure to abrocitinib and the development of malignancies. Long-term safety evaluations are ongoing.
The risks and benefits of abrocitinib treatment should be considered prior to initiating in patients with a known malignancy other than a successfully treated NMSC or cervical cancer in situ or when considering continuing abrocitinib therapy in patients who develop a malignancy. Periodic skin examination is recommended for patients who are at increased risk for skin cancer.
Haematologic abnormalities
Confirmed ALC < 0.5 × 103/mm3 and platelet count < 50 × 103/mm3 were observed in less than 0.5% of patients in clinical studies. Treatment with abrocitinib should not be initiated in patients with a platelet count < 150 × 103/mm3, an ALC < 0.5 × 103/mm3, an ANC < 1 × 103/mm3 or who have a haemoglobin value < 8 g/dL (see section 4.2). Platelet count and ALC should be monitored 4 weeks after initiation of therapy with abrocitinib and thereafter according to routine patient management. (see section 4.8).
Lipids
Dose-dependent increases in blood lipid parameters were reported in patients treated with abrocitinib (see section 4.8). Lipid parameters should be assessed 4 weeks following initiation of abrocitinib therapy. Patients who develop changes in lipid parameters should be further monitored and managed according to clinical guidelines, due to the known cardiovascular risks associated with dyslipidaemia. The effect of these drug-induced changes in lipid parameters on cardiovascular morbidity and mortality has not been determined.
Laboratory monitoring
Table 2. Laboratory measures initiation and monitoring guidance
| Laboratory measure | Monitoring guidance | Action |
|---|---|---|
| Platelet counts | Before treatment initiation, 4 weeks after initiation and thereafter according to routine patient management. | Treatment should be discontinued if platelet counts are <50 × 103/mm3. |
| Absolute Lymphocyte Count (ALC) | Before treatment initiation, 4 weeks after initiation and thereafter according to routine patient management. | Treatment should be interrupted if ALC is < 0.5 × 103/mm3 and may be restarted once ALC returns above this value. Treatment should be discontinued if confirmed. |
| Lipid parameters | Before treatment initiation, 4 weeks after initiation and thereafter according to clinical guidelines for hyperlipidaemia. | Patients should be monitored according to clinical guidelines for hyperlipidaemia. |
Excipients
Lactose Patients with rare hereditary problems of galactose intolerance, total lactase deficiency or glucose galactose malabsorption should not take this medicinal product.
Sodium content This medicinal product contains less than 1 mmol sodium (23 mg) per tablet. Patients on low sodium diets can be informed that this medicinal product is essentially ‘sodium-free’.
4.5 Interaction with other medicinal products and other forms of interaction
Potential for other medicines to affect pharmacokinetics of abrocitinib
Abrocitinib is metabolised predominantly by CYP2C19 and CYP2C9 enzymes, and its active metabolites are renally excreted and are substrates of the organic anion transporter 3 (OAT3). Therefore, exposures of abrocitinib and/or its active metabolites may be affected by medicinal products that strongly inhibit or induce CYP2C19 or CYP2C9 or inhibit the OAT3 transporter. Dose adjustments, as appropriate, based on these results are outlined in section 4.2.
Co-administration with CYP2C19/CYP2C9 inhibitors
When abrocitinib 100 mg was administered concomitantly with fluvoxamine (a strong CYP2C19 and moderate CYP3A inhibitor) or fluconazole (a strong CYP2C19, moderate CYP2C9 and CYP3A inhibitor), the extent of exposure of abrocitinib active moiety (see section 5.2) increased by 91% and 155%, respectively, compared with administration alone.
Co-administration with CYP2C19/CYP2C9 inducers
Administration of abrocitinib 200 mg after multiple doses with rifampin, a strong inducer of CYP enzymes, resulted in reduction of abrocitinib active moiety exposures by approximately 56%.
Co-administration with OAT3 inhibitors
When abrocitinib 200 mg was administered concomitantly with probenecid, an OAT3 inhibitor, abrocitinib active moiety exposures increased by approximately 66%. This is not clinically significant, and a dose adjustment is not needed.
Potential for abrocitinib to affect pharmacokinetics of other medicines
In vitro, abrocitinib or its metabolites were not significant inhibitors or inducers of CYPs (CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4) or of uridine diphosphate glucuronyltransferases (UGTs) (UGT1A1, UGT1A4, UGT1A6, UGT1A9, and UGT2B7). In vitro, abrocitinib is an inhibitor of P glycoprotein (P-gp), OAT3, organic cation transporter (OCT)1, multidrug and toxin compound extrusion protein (MATE)1/2K and breast cancer resistance protein (BCRP) but is not an inhibitor of organic anion transporting polypeptide (OATP) 1B1/1B3, bile salt export pump (BSEP), OAT1 or OCT2 at clinically meaningful concentrations. The metabolites do not change the transporter inhibition risk compared to abrocitinib.
No clinically significant effects of abrocitinib were observed in drug interaction studies with oral contraceptives (e.g., ethinyl oestradiol/levonorgestrel), or with substrates of BCRP and OAT3 (e.g., rosuvastatin), MATE1/2K (e.g., metformin) and CYP3A4 (e.g., midazolam). Coadministration of dabigatran etexilate (a P-gp substrate), with a single dose of abrocitinib 200 mg increased dabigatran AUCinf and Cmax by approximately 53% and 40%, respectively, compared with administration alone. The effect of abrocitinib on pharmacokinetics of digoxin, a P-gp substrate, has not been evaluated. Caution should be exercised as the levels of digoxin may increase.
4.6 Fertility, pregnancy and lactation
Pregnancy
There are no or limited amount of data on the use of abrocitinib in pregnant women. Studies in animals have shown reproductive toxicity. Abrocitinib has been shown to cause skeletal variations in pregnant rats and rabbits and to affect parturition and peri/postnatal development in pregnant rats and rabbits and to affect parturition and peri/postnatal development in pregnant rats (see section 5.3).
Abrocitinib is contraindicated during pregnancy (see section 4.3).
Women of childbearing potential
Women of reproductive potential should be advised to use effective contraception during treatment with abrocitinib and for 1 month following the final dose of abrocitinib. Consider pregnancy planning and prevention for females of reproductive potential.
Breast-feeding
There are no data on the presence of abrocitinib in human milk, the effects on the breast fed infant, or the effects on milk production. Abrocitinib was secreted in milk of lactating rats. A risk to newborns/infants cannot be excluded and abrocitinib should not be used during breast feeding.
Fertility
Based on the findings in rats, oral administration of abrocitinib may result in temporary reduced fertility in females of reproductive potential. These effects on female rat fertility were reversible 1 month after cessation of abrocitinib oral administration (see section 5.3).
4.7 Effects on ability to drive and use machines
Abrocitinib has no or negligible sedating effect. However, patients who experience dizziness after the intake of abrocitinib should refrain from driving or using machines.
4.8 Undesirable effects
Summary of safety profile
The most commonly reported adverse reactions occurring in ≥2% of patients treated with abrocitinib in placebo-controlled studies were nausea (10.3%), headache (6.8%), herpes simplex (3.8%), acne (3.2%), blood creatine phosphokinase increased (2.6%), dizziness (2.3%), and vomiting (2.3%). The most frequent serious adverse reactions in atopic dermatitis patients were infections (see section 4.4).
Tabulated list of adverse reactions
A total of 2,856 patients were treated with abrocitinib in clinical studies in atopic dermatitis representing 1,614 patient-years of exposure. There were 606 patients with more than 1 year of exposure to abrocitinib. Four placebo-controlled studies were integrated (608 patients on 100 mg once daily, 590 patients on 200 mg once daily and 342 patients on placebo) to evaluate the safety of abrocitinib in comparison to placebo for up to 16 weeks.
Table 3. Adverse reactions
| System Organ Class | Very Common | Common | Uncommon |
|---|---|---|---|
| Infections and infestations | Herpes simplex (a) | Herpes zoster (b) | |
| Blood and lymphatic system disorders | Thrombocytopenia Lymphopenia | ||
| Metabolism and nutrition disorders | Hyperlipidaemia (c) | ||
| Nervous system disorders | Headache Dizziness | ||
| Vascular disorders | Venous thromboembolism (d) | ||
| Gastrointestinal disorders | Nausea | Vomiting Abdominal pain upper | |
| Skin and subcutaneous tissue disorders | Acne | ||
| Investigations | Creatine phosphokinase elevations increased |
a. Herpes simplex includes oral herpes, ophthalmic herpes simplex, genital herpes, and eczema herpeticum.
b. Herpes zoster includes ophthalmic herpes zoster.
c. Hyperlipidaemia included dyslipidaemia and hypercholesterolaemia.
d. Venous thromboembolism includes pulmonary embolism and deep vein thrombosis.
Listed in Table 3 are adverse reactions observed in atopic dermatitis clinical trials presented by system organ class and frequency, using the following categories: very common (≥1/10), common (≥1/100 to <1/10), uncommon (≥1/1,000 to <1/100), rare (≥1/10,000 to <1/1,000), very rare (<1/10,000). Within each frequency grouping, adverse events are presented in order of decreasing seriousness.
Description of selected adverse reactions
Overall Infections In placebo-controlled studies, for up to 16 weeks, overall infections have been reported by 26.3% of patients treated with placebo and by 35.2% and 34.6% of patient treated with abrocitinib 100 mg and 200 mg, respectively. Most infections were mild or moderate.
Serious infections In placebo controlled studies, for up to 16 weeks, serious infections have been reported in 2 patients (2.31 per 100 patient-years) treated with placebo, 6 patients (3.80 per 100 patient-years) treated with abrocitinib 100 mg, and 2 patients (1.28 per 100 patient-years) treated with abrocitinib 200 mg. Among all patients treated with abrocitinib, including the long-term extension study, serious infections were reported in 17 patients (2.65 per 100 patient-years) treated with abrocitinib 100 mg and 24 patients (2.33 per 100 patient-years) treated with abrocitinib 200 mg. The most commonly reported serious infections were herpes simplex, herpes zoster, and pneumonia (see section 4.4).
Opportunistic infections All opportunistic infections were cases of multidermatomal cutaneous herpes zoster. Among all patients treated with abrocitinib, including the long-term extension study, opportunistic infections were reported in 1 patient (0.16 per 100 patient-years) treated with abrocitinib 100 mg and 9 patients (0.87 per 100 patient-years) treated with abrocitinib 200 mg. Most cases of opportunistic herpes zoster were mild or moderate.
Herpes zoster In placebo controlled studies, for up to 16 weeks, herpes zoster infections (including serious and opportunistic infections noted above) have been reported in 0 patients treated with placebo, 3 patients (1.9 per 100 patient-years) treated with abrocitinib 100 mg, and 8 patients (5.16 per 100 patient-years) treated with abrocitinib 200 mg. Among all patients treated with abrocitinib, including the long-term extension study, herpes zoster infections were reported in 13 patients (2.04 per 100 patient-years) treated with abrocitinib 100 mg and 44 patients (4.34 per 100 patient-years) treated with abrocitinib 200 mg. The incidence rate of herpes zoster in patients with severe atopic dermatitis at baseline (4.93 per 100 patient-years) was higher than that of patients with moderate disease at baseline (2.49 per 100 patient years). The incidence rate of herpes zoster in patients 65 years of age and older treated with abrocitinib was higher than that of younger patients (see Elderly section below).
Venous thromboembolism Among all patients treated with abrocitinib, including the long term extension study, PE was reported in 3 patients (0.18 per 100 patient-years), all treated with abrocitinib 200 mg. Events of DVT were reported in 2 patients (0.09 per 100 patient-years) treated with abrocitinib 200 mg (see section 4.4).
Thrombocytopenia In placebo controlled studies, for up to 16 weeks, treatment with abrocitinib was associated with a dose-related decrease in platelet count. Maximum effects on platelets were observed within 4 weeks, after which the platelet count returned towards baseline despite continued therapy. Confirmed platelet counts of <50 × 103/mm3 were reported in 1 patient (0.1%) exposed to abrocitinib 200 mg, 0 patients treated with abrocitinib 100 mg or placebo. Among all patients exposed to abrocitinib, including the long-term extension study, confirmed platelet counts of < 50 × 103/mm3 were reported in 2 patients (0.1%), both treated with abrocitinib 200 mg (see section 4.4).
Lymphopenia In placebo controlled studies, for up to 16 weeks, confirmed ALC < 0.5 × 103/mm3 occurred in 2 patients (0.3%) treated with abrocitinib 200 mg and 0 patients treated with abrocitinib 100 mg or placebo. Both cases occurred in the first 4 weeks of exposure. Among all patients exposed to abrocitinib, including the long-term extension, confirmed ALC < 0.5 × 103/mm3 were reported in 4 patients (0.1%) treated with 200 mg of abrocitinib and 0 patients treated with abrocitinib 100 mg (see section 4.4).
Lipid elevations In placebo controlled studies, for up to 16 weeks, there was a dose-related percent increase in low density lipoprotein cholesterol (LDL-c), total cholesterol, and high-density lipoprotein cholesterol (HDL-c) relative to placebo at Week 4 which remained elevated through the final visit in the treatment period. There was no change in the LDL/HDL ratio or triglycerides. Events related to hyperlipidaemia occurred in 1 patient (0.2%) exposed to abrocitinib 100 mg, 7 patients (1.2%) exposed to abrocitinib 200 mg and 0 patients exposed to placebo (see section 4.4).
Creatine phosphokinase elevations (CPK) In placebo controlled studies, for up to 16 weeks, events of blood CPK increased were reported in 1.5% of patients treated with placebo, 2.3% and 2.9% of patients treated with 100 mg and 200 mg of abrocitinib, respectively. Most elevations were transient, and none led to discontinuation. In the clinical studies, there were no reported events of rhabdomyolysis.
Nausea Nausea was most frequent in the first week of abrocitinib therapy and generally resolved with continued therapy. The median duration of nausea was 15 days. Most of the cases were mild to moderate in severity.
Paediatric population
The pharmacokinetics, safety and efficacy of abrocitinib in paediatric patients under 12 years of age have not yet been established.
Of the 2,856 patients with atopic dermatitis exposed to abrocitinib, a total of 364 adolescents (12 to ˂18 years of age) were enrolled in abrocitinib studies. The safety profile observed in adolescents in atopic dermatitis clinical studies was similar to that of the adult population. There were no adolescent patients who developed platelet counts < 75 x 103/mm3 or ALC < 0.5 x 103/mm3.
Elderly
A total of 145 patients 65 years of age and older were enrolled in abrocitinib studies. The safety profile observed in elderly patients was similar to that of the adult population overall. A higher proportion of patients 65 years of age and older discontinued from clinical studies compared to younger patients. Among all patients exposed to abrocitinib, including the long-term extension study, confirmed ALC < 0.5 × 103/mm3 occurred only in patients 65 years of age and older. A higher proportion of patients 65 years of age and older had platelet counts < 75 × 103/mm3. The incidence rate of herpes zoster in patients 65 years of age and older treated with abrocitinib (7.40 per 100 patient-years) was higher than that of patients 18 to less than 65 years of age (3.44 per 100 patient years) and less than 18 years of age (2.12 per 100 patient years). There is limited data in patients above 75 years of age.
Reporting of suspected adverse reactions
For reporting suspected adverse reactions during the EAMS, healthcare professionals are requested to report any suspected adverse reactions via the Compassionate Use Adverse Event form to Pfizer within 24 hours of awareness of the event for assessment and processing. The contact details for the Pfizer Drug Safety Unit are: GBR.AEReporting@pfizer.com.
4.9 Overdose
Abrocitinib was administered in clinical studies up to a single oral dose of 800 mg. There is no experience with overdose of abrocitinib. There is no specific antidote for overdose with abrocitinib. In case of an overdose, it is recommended that the patient be monitored for signs and symptoms of adverse reactions. Treatment should be symptomatic and supportive.
Pharmacokinetic data up to and including a single oral dose of 800 mg in healthy adult volunteers indicate that more than 90% of the administered dose is expected to be eliminated within 48 hours.
5. Pharmacological properties
5.1 Pharmacodynamic properties
Mechanism of action
Abrocitinib is a Janus kinase (JAK)1 inhibitor. JAKs are intracellular enzymes which transmit signals arising from cytokine or growth factor-receptor interactions on the cellular membrane to influence cellular processes of haematopoiesis and immune cell function. Within signalling pathways, JAKs phosphorylate and activate Signal Transducers and Activators of Transcription (STATs) which modulate intracellular activity including gene expression. Abrocitinib modulates the signalling pathway at the point of JAK1, preventing the phosphorylation and activation of STATs.
Abrocitinib reversibly and selectively inhibits JAK1 by blocking the adenosine triphosphate (ATP) binding site. In a cell-free isolated enzyme assay, abrocitinib has biochemical selectivity for JAK1 over the other 3 JAK isoforms JAK2 (28 fold), JAK3 (> 340-fold) and tyrosine kinase (TYK) 2 (43-fold), and even higher selectivity over the broader kinome. In cellular settings, where JAK enzymes transmit signals in pairs (i.e., JAK1/JAK2, JAK1/JAK3, JAK1/TYK2, JAK2/JAK2, JAK2/TYK2), abrocitinib preferentially inhibits cytokine-induced STAT phosphorylation mediated by receptors utilising JAK1 relative to receptors utilising JAK2 only or JAK2/TYK2 pairs. The relevance of inhibition of specific JAK enzymes to therapeutic effectiveness is not currently known. Both the parent compound and the active metabolites inhibit cytokine signalling with similar levels of selectivity.
Pharmacodynamic effects
Treatment with abrocitinib was associated with dose-dependent reduction in serum markers of inflammation, including high sensitivity C-reactive protein (hsCRP), interleukin-31 (IL 31) and thymus and activation-regulated chemokine (TARC). These changes returned to near baseline within 4 weeks of drug discontinuation.
Clinical efficacy and safety
The efficacy and safety of abrocitinib as monotherapy and in combination with background medicated topical therapies were evaluated in 3 pivotal randomised, double-blind, placebo-controlled studies (MONO-1, MONO 2, and COMPARE) in 1,616 patients 12 years of age and older with moderate-to-severe atopic dermatitis as defined by Investigator’s Global Assessment (IGA) score ≥ 3, Eczema Area and Severity Index (EASI) score ≥ 16, body surface area (BSA) involvement ≥ 10%, and Peak Pruritus Numerical Rating Scale (PP-NRS) ≥ 4 at the baseline visit prior to randomisation.
Patients in these studies were those who had inadequate response to previous topical medication or were patients for whom topical treatments were medically inadvisable, or who had received systemic therapies including dupilumab. In each of the pivotal studies, over 40% of patients had prior exposure to systemic therapy. In MONO-1 and MONO-2, 6% of the patients had received dupilumab, whereas prior use of dupilumab was not allowed in COMPARE.
Eligible patients from qualifying parent studies were able to enrol in the long-term extension study EXTEND, e.g. if they completed the full treatment period of the any of the pivotal qualifying parent studies.
MONO-1, MONO-2, and COMPARE assessed the co-primary endpoints of IGA and EASI 75 responses at Week 12. Key secondary endpoints in MONO-1 and MONO-2 included improvement of ≥4 points in the severity of PP-NRS (PP-NRS4) at Week 12 and change from baseline to Week 12 for the Pruritus and Symptoms Assessment for Atopic Dermatitis (PSAAD). The PSAAD is an 11-item, self-reported instrument using a 24-hour recall period, designed to assess the severity of key symptoms and signs of atopic dermatitis including itching, pain, dryness, flaking, cracking, bumps, redness, discolouration, bleeding, fluid, and swelling. Key secondary endpoints in COMPARE were PP-NRS4 at Week 2 in addition to IGA response and EASI-75 at Week 16. The designs of the pivotal and long-term extension studies are summarised in Table 4.
Table 4. Clinical trial summary
| Study name (regimen type)/Treatment duration | Population (number of randomised patients) | Treatment arms | Primary and key secondary endpoints |
|---|---|---|---|
| MONO-1 (monotherapy)/12 weeks | Adults and adolescents (387) | Abrocitinib 200 mg OD, Abrocitinib 100 mg OD, Placebo | Co-primary: IGA response (a) at week 12, EASI-75 (b) at week 12. Key secondary: PP-NRS4 (c) at Weeks 2, 4 and 12, Change from baseline in PSAAD (d) at Week 12 |
| MONO-2 (monotherapy)/12 weeks | Adults and adolescents (391) | Abrocitinib 200 mg OD, Abrocitinib 100 mg OD, Placebo | Co-primary: IGA response at week 12, EASI-75 at week 12. Key secondary: PP-NRS4 at Weeks 2, 4 and 12, Change from baseline in PSAAD at Week 12 |
| COMPARE (combination therapy)/16 weeks | Adults (838) | Abrocitinib 200 mg OD, Abrocitinib 100 mg OD, Placebo, Dupilumab 300 mg Q2W SC (e). All patients received background medicated topical therapy. | Co-primary: IGA response at week 12, EASI-75 at week 12. Key secondary: PP-NRS4 at Week 2, IGA response at week 16, EASI-75 at week 16 |
| EXTEND (long-term extension) variable | Adults and adolescents (approx. 2,300) | Abrocitinib 200 mg OD, Abrocitinib 100 mg OD | Primary: Long-term safety |
Primary and key secondary endpoints were controlled for multiplicity. Abbreviations: EASI=Eczema Area and Severity Index; IGA=Investigator’s Global Assessment; PP-NRS=Peak Pruritus Numerical Rating Scale; PSAAD = Pruritus and Symptoms Assessment for Atopic Dermatitis; OD=once daily; Q2W=once every 2 weeks; SC=subcutaneously.
a. IGA response was based on IGA score of clear (0) or almost clear (1) (on a 5 point scale) and a reduction from baseline of ≥ 2 points.
b. EASI-75 was based on ≥ 75% improvement in EASI from baseline.
c. PP-NRS4 response was based on an improvement of ≥ 4 points in the severity of PP-NRS.
d. PSAAD ranges from 0 to 10 with higher scores indicating greater levels of atopic dermatitis symptom severity.
e. Dupilumab treatment in COMPARE: An initial dose of 600 mg on Day 1, followed by 300 mg Q2W.
Clinical response Treatment with abrocitinib 100 mg or 200 mg once daily as monotherapy or in combination with background medicated topical therapy resulted in improvement in objective signs of atopic dermatitis and patient-reported pruritus.
Monotherapy studies In both pivotal monotherapy studies (MONO-1, MONO-2), the proportion of patients who achieved IGA and/or EASI-75 response was significantly higher in patients who received abrocitinib 100 mg or 200 mg once daily compared with placebo at Week 12 (see Table 5).
A significantly higher proportion of patients who achieved PP-NRS4 (defined as an improvement of ≥ 4 points in the severity of PP-NRS) with abrocitinib 100 mg or 200 mg once daily compared with placebo was observed as soon as Week 2 and persisting through Week 12. Higher proportions of patients achieved PP-NRS4 with abrocitinib 100 mg or 200 mg once daily compared with placebo by day 6 and day 3 (2 days after the first dose), respectively (see Table 5).
Table 5. Efficacy results of abrocitinib monotherapy at Week 12
MONO-1
| ABR | ABR | ||
|---|---|---|---|
| 200 mg OD N=154 | 100 mg OD N=154 | Placebo N=77 | |
| % Responders (95% CI) | % Responders (95% CI) | % Responders (95% CI) | |
| IGA 0 or 1 (a) | 43.8 (g) (35.9, 51.7) | 23.7 (e) (17.0, 30.4) | 7.9 (1.8, 14.0) |
| EASI-50 (b) | 75.8 (k) (69.0, 82.6) | 57.7 (k) (49.9, 65.4) | 22.4 (13.0, 31.7) |
| EASI-75 (b) | 62.7 (g) (55.1, 70.4) | 39.7 (g) (32.1, 47.4) | 11.8 (4.6, 19.1) |
| EASI-90 (b) | 38.6 (k) (30.8, 46.3) | 18.6 (i) (12.5, 24.7) | 5.3 (0.2, 10.3) |
| EASI-100 (b) | 13.1 (i) (7.7, 18.4) | 6.4 (h) (2.6, 10.3) | 0 (0.0, 4.7) |
| PP-NRS4 (c, d) | 57.2 (g) (48.8, 65.6) | 37.7 (f) (29.2, 46.3) | 15.3 (6.6, 24.0) |
| PP-NRS (0 or 1) | 35.4 (k) (27.2, 43.6) | 21.1 (i) (13.9, 28.4) | 3.2 (0.0, 7.5) |
| % Change from baseline (95% CI) | % Change from baseline (95% CI) | % Change from baseline (95% CI) | |
| LSM EASI | -73.5 (k) (-79.1, -68.0) | -57.5 (k) (-63.1, -51.9) | -28.4 (-36.5, -20.3) |
| LSM PP-NRS | -56.5 (k) (-63.6, -49.5) | -39.5 (i) (-46.7, -32.3) | -19.5 (-30.0, -9.0) |
| LSM SCORAD | -55.1(k) (-60.1, -50.2) | -41.5 (k) (-46.5, -36.5) | -21.6 (-28.7, -14.5) |
| Change from baseline (95% CI) | Change from baseline (95% CI) | Change from baseline (95% CI) | |
| LSM PSAAD | -3.2 (g) (-3.6, -2.8) | -2.2 (e) (-2.6, -1.9) | -1.1 (-1.7, -0.6) |
MONO-2
| ABR | ABR | ||
|---|---|---|---|
| 200 mg OD N=155 | 100 mg OD N=158 | Placebo N=78 | |
| % Responders (95% CI) | % Responders (95% CI) | % Responders (95% CI) | |
| IGA 0 or 1 (a) | 38.1 (g) (30.4, 45.7) | 28.4 (f) (21.3, 35.5) | 9.1 (2.7, 15.5) |
| EASI-50 (b) | 79.9 (k) (73.5, 86.2) | 68.4 (k) (61.1, 75.7) | 19.5 (10.6, 28.3) |
| EASI-75 (b) | 61.0 (g) (53.3, 68.7) | 44.5 (g) (36.7, 52.3) | 10.4 (3.6, 17.2) |
| EASI-90 (b) | 37.7 (k) (30.0, 45.3) | 23.9 (k) (17.2, 30.6) | 3.9 (0.0, 8.2) |
| EASI-100 (b) | 7.1 (h) (3.1, 11.2) | 5.2 (h) (1.7, 8.6) | 0 (0.0, 4.7) |
| PP-NRS4 (c, d) | 55.3 (g) (47.2, 63.5) | 45.2 (g) (37.1, 53.3) | 11.5 (4.1, 19.0) |
| PP-NRS (0 or 1) | 32.4 (k) (24.5, 40.2) | 21.3 (i) (14.5, 28.0) | 5.5 (0.3, 10.7) |
| % Change from baseline (95% CI) | % Change from baseline (95% CI) | % Change from baseline (95% CI) | |
| LSM EASI | -73.3 (k) (-79.7, -66.9) | -60.0 (k) (-66.5, -53.6) | -28.6 (-38.4, -18.8) |
| LSM PP-NRS | -56.9 (k) (-64.0, -49.8) | -43.5 (j) (-50.7, -36.3) | -20.8 (-31.6, -9.9) |
| LSM SCORAD | -56.2 (k) (-61.2, -51.1) | -45.8 (k) (-50.9, -40.7) | -22.7 (-30.4, -15.1) |
| Change from baseline (95% CI) | Change from baseline (95% CI) | Change from baseline (95% CI) | |
| LSM PSAAD | -3.0 (g) (-3.3, -2.7) | -2.4 (g) (-2.8, -2.1) | -0.8 (-1.3, -0.3) |
Abbreviations: ABR=abrocitinib; CI=confidence interval; EASI=Eczema Area and Severity Index; LSM=least squares mean; IGA=Investigator Global Assessment; N=number of patients randomised; PP-NRS=Peak Pruritus Numerical Rating Scale; PSAAD=Pruritus and Symptoms Assessment for Atopic Dermatitis; OD=once daily; SCORAD=SCORing Atopic Dermatitis.
a. IGA responders were patients with IGA score of clear (0) or almost clear (1) (on a 5 point scale) and a reduction from baseline of 2 points.
b. EASI-50, -75, -90 and -100 responders were patients with ≥ 50%, ≥ 75%, ≥ 90%, and ≥ 100% improvement, respectively, in EASI, from baseline.
c. The proportion of PP-NRS4 responders was also significantly higher with abrocitinib 200 mg and 100 mg once daily than placebo at Week 2, Week 4, and Week 8 in both MONO-1 and MONO-2.
d. PP-NRS4 responders were patients with ≥ 4-point improvement in PP-NRS from baseline.
e. Multiplicity controlled p <0.01 versus placebo.
f. Multiplicity controlled p < 0.001 versus placebo.
g. Multiplicity controlled p < 0.0001 versus placebo.
h. Nominal p < 0.05 versus placebo.
i. Nominal p < 0.01 versus placebo.
j. Nominal p < 0.001 versus placebo.
k. Nominal p < 0.0001 versus placebo.
The proportion of patients who achieved EASI-90 or PP-NRS4 over time in studies MONO-1 and MONO-2 are shown in Figures 1 and 2.
Figure 1. Proportion of patients who achieved EASI-90 over time in MONO-1 and MONO-2
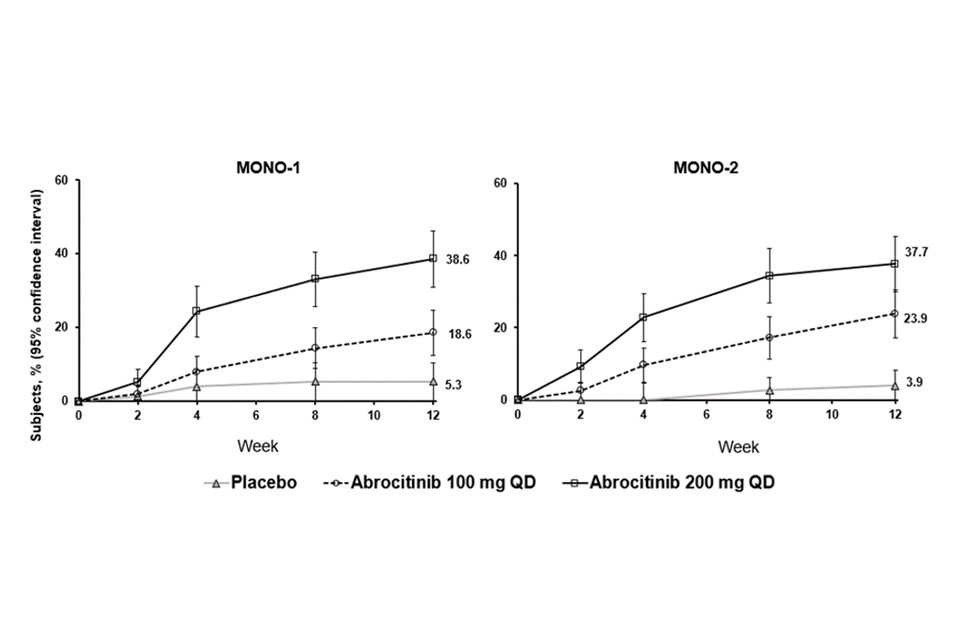
Abbreviations: EASI=Eczema Area and Severity Index; QD=once daily. EASI-90 was based on EASI ≥90% improvement from baseline.
Figure 2. Proportion of patients who achieved PP-NRS4 over time in MONO-1 and MONO-2
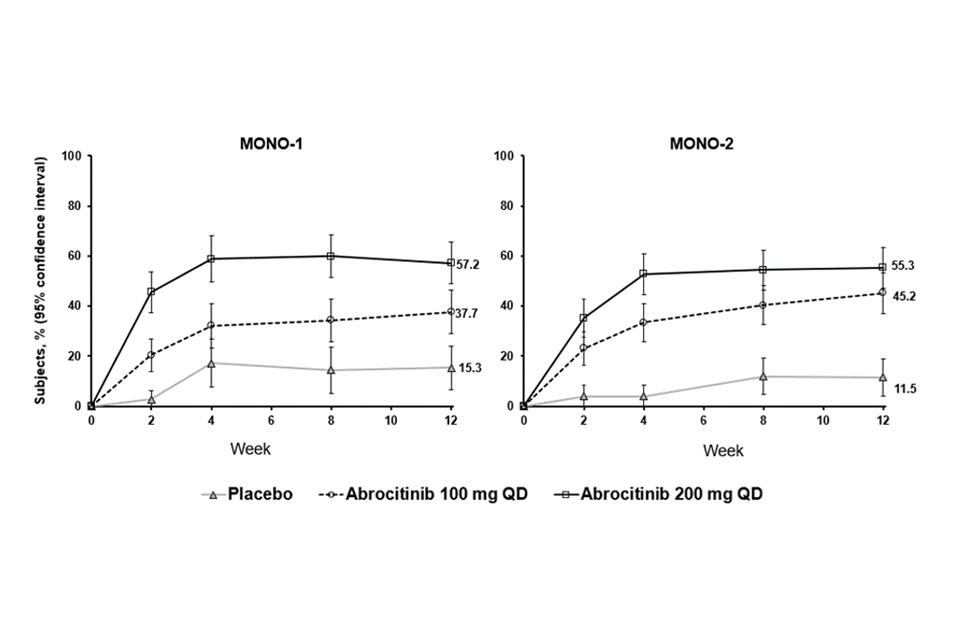
Abbreviations: PP-NRS=Peak Pruritus Numerical Rating Scale; QD=once daily. PP-NRS4 responders were patients with ≥4-point improvement in Peak Pruritis Numerical Rating Scale (PP-NRS) from baseline.
Treatment effects in subgroups (e.g. weight, age, sex, race and prior systemic immunosuppressant treatment) in MONO-1 and MONO-2 were consistent with the results in the overall study population.
Combination therapy study In the pivotal combination therapy study (COMPARE), the proportion of patients who achieved IGA or EASI-75 response was significantly higher in patients who received abrocitinib 100 mg or 200 mg once daily compared with placebo at Week 12 (see Table 6).
The proportions of patients achieving PP-NRS4 with abrocitinib 100 mg and 200 mg once daily were significantly higher than placebo by Day 9 and Day 4, respectively, and remained significantly higher than placebo with both abrocitinib doses at Week 2 and Week 16.
The proportion of patients achieving PP-NRS4 with abrocitinib 200 mg once daily was significantly higher than dupilumab as early as Day 4 and remained significantly higher than dupilumab at Week 2. The proportion of patients achieving PP-NRS4 was similar between abrocitinib 100 mg once daily and dupilumab at Week 2.
Table 6. Efficacy results of abrocitinib with concomitant topical therapy at Week 12
| Week 2 | Week 2 | Week 2 | Week 2 | Week 12 | Week 12 | Week 12 | Week 12 | Week 16 | Week 16 | Week 16 | Week 16 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Abrocitinib | Abrocitinib | Abrocitinib | Abrocitinib | Abrocitinib | Abrocitinib | |||||||
| % Responders | % Responders | % Responders | % Responders | % Responders | % Responders | % Responders | % Responders | % Responders | % Responders | % Responders | % Responders | |
| IGA 0 or 1 (a) | 18.4 (i) | 15.2 (h) | 6.3 | 4.7 | 48.4 (e) | 36.6 (e) | 14.0 | 36.5 | 47.5 (e) | 34.8 (e) | 12.9 | 38.8 |
| EASI-50 (b) | 60.5 (j) | 53.1 (j) | 21.9 | 35.7 | 86.3 (j) | 75.3 (j) | 52.7 | 80.9 | 87.3 (j) | 81.2 (j) | 57.3 | 84.1 |
| EASI-75 (b) | 30.0 (j) | 25.4 (i) | 10.9 | 14.0 | 70.3 (e) | 58.7 (e) | 27.1 | 58.1 | 71.0 (e) | 60.3 (e) | 30.6 | 65.5 |
| EASI-90 (b) | 11.2 (h) | 8.3 (g) | 2.3 | 2.6 | 46.1 (j) | 36.6 (j) | 10.1 | 34.9 | 48.9 (j) | 38.0 (j) | 11.3 | 38.8 |
| EASI-100 (b) | 4.5 (g) | 1.3 | 0 | 0.4 | 12.3 (i) | 8.1 (h) | 1.6 | 6.6 | 13.6 (h) | 12.7 (h) | 4.0 | 5.2 |
| PP-NRS4 (c) | 49.1 (e, f) | 31.8 (d) | 13.8 | 26.4 | 63.1 (j) | 47.5(i) | 28.9 | 54.5 | 62.8 (j) | 47.0 (h) | 28.7 | 57.1 |
| PP-NRS (0 or 1) | 15.0 (h) | 8.9 | 4.6 | 4.6 | 36.9 (j) | 21.1 (i) | 7.4 | 24.9 | 32.0 (i) | 24.7 (g) | 11.7 | 24.2 |
| % Change from baseline | % Change from baseline | % Change from baseline | % Change from baseline | % Change from baseline | % Change from baseline | % Change from baseline | % Change from baseline | % Change from baseline | % Change from baseline | % Change from baseline | % Change from baseline | |
| LSM EASI | -54.6 (j) | -49.3 (j) | -21.2 | -38.8 | -80.6 (j) | -73.8 (j) | -47.7 | -75.4 | -83.2 (j) | -75.2 (j) | -53.8 | -80.2 |
| LSM PP-NRS | -45.6 (j) | -35.5 (j) | -19.5 | -29.3 | -63.3 (j) | -48.2 (j) | -30.4 | -54.8 | -64.1 (j) | -49.1 (j) | -30.3 | -58.5 |
| LSM SCORAD | -41.7 (j) | -34.6 (j) | -18.1 | -27.7 | -65.2 (j) | -54.2 (j) | -33.5 | -58.4 | -65.4 (j) | -55.6 (j) | -38.8 | -61.9 |
| LSM PSAAD | -2.3 (j) | -1.8 (j) | -0.9 | -1.6 | -3.6 (j) | -2.7 (j) | -1.6 | -3.2 | -3.6 (j) | -2.8 (j) | -1.7 | -3.4 |
Abbreviations: DUP=dupilumab; EASI=Eczema Area and Severity Index; LSM=least squares mean; N=number of patients randomized; PBO=placebo; PP-NRS=Peak Pruritus Numerical Rating Scale; PSAAD=Pruritus and Symptoms Assessment for Atopic Dermatitis; SCORAD=SCORing Atopic Dermatitis.
a. IGA responder was a patient with IGA score of clear (0) or almost clear (1) (on a 5 point scale) and a reduction from baseline of ≥ 2 points.
b. EASI-50, -75, -90 and -100 responders were patients with ≥ 50%, ≥ 75%, ≥ 90% and ≥ 100% improvement in EASI, respectively, from baseline.
c. PP-NRS4 response was based on an improvement of ≥ 4 points in the severity of PP-NRS.
d. Multiplicity-controlled p < 0.001 versus placebo.
e. Multiplicity-controlled p < 0.0001 versus placebo.
f. Multiplicity-controlled p < 0.0001 versus. dupilumab. Statistical comparison between either abrocitinib dose and dupilumab was only performed on the proportion of patients achieving PP-NRS4 at Week 2.
g. Nominal p < 0.05 versus. placebo.
h. Nominal p < 0.01 versus placebo.
i. Nominal p < 0.001 versus placebo.
j. Nominal p < 0.0001 versus placebo.
The proportion of patients who achieved EASI-90 or PP-NRS4 over time in COMPARE are shown in Figure 3.
Figure 3. Proportion of patients who achieved A) EASI-90 and B) PP-NRS4 over time in COMPARE
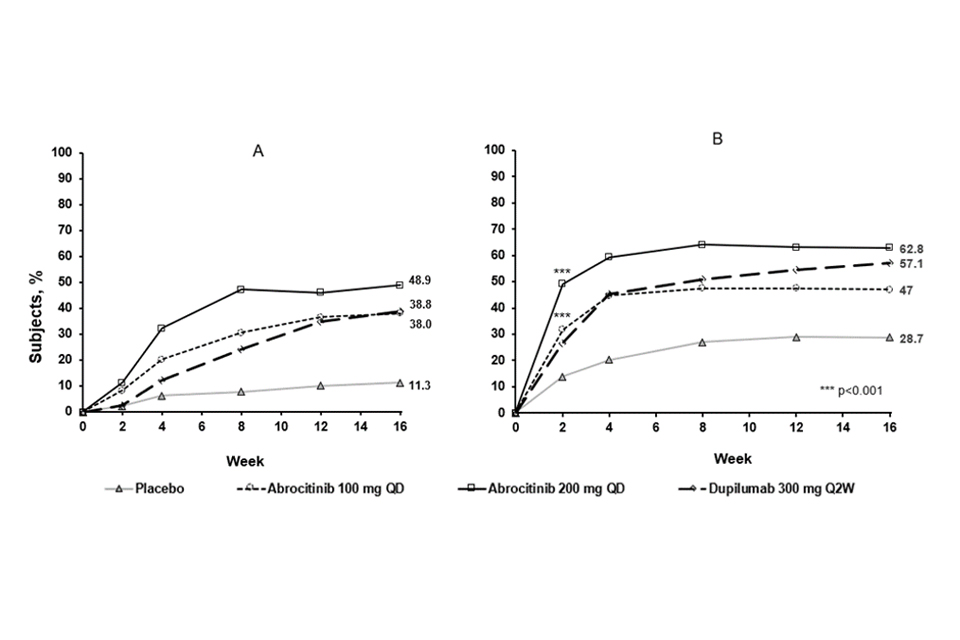
Abbreviations: EASI=Eczema Area and Severity Index; PP-NRS=Peak Pruritus Numerical Rating Scale; QD=once daily; Q2W=every 2 weeks. EASI 90 was based on EASI ≥90% improvement from baseline. PP-NRS4 responders were patients with ≥4-point improvement in Peak Pruritis Numerical Rating Scale (PP-NRS) from baseline.
Treatment effects in subgroups (e.g. weight, age, sex, race and prior immunosuppressant treatment) in COMPARE were consistent with the results in the overall study population.
Late-onset efficacy Eligible patients who completed the full treatment period of a qualifying parent study (e.g., MONO-1, MONO 2, COMPARE) were considered for enrollment in the long-term extension study EXTEND, which allows patients to extend abrocitinib treatment for at least 92 weeks or until availability of commercial product in their country. In EXTEND, patients received abrocitinib with or without background medicated topical therapy. Patients who were previously randomised to abrocitinib EEIG 100 mg or 200 mg once daily in qualifying studies continued the same dose in EXTEND as in the parent study, and the blind was maintained. Patients not previously randomised to abrocitinib in a qualifying parent study were randomised to either abrocitinib 100 mg or 200 mg once daily upon entering EXTEND.
Among all patients who did not achieve IGA (0 or 1) response after 12 weeks of abrocitinib treatment and entered EXTEND, 14% and 25% of patients continuing abrocitinib 100 mg once daily in EXTEND achieved IGA (0 or 1) response by Week 16 and Week 24 (with 4 and 12 additional weeks of treatment), respectively, and 19% and 29% of patients continuing abrocitinib 200 mg once daily achieved IGA response by Week 16 and Week 24, respectively. Among all patients who did not achieve EASI-75 after 12 weeks of abrocitinib treatment and entered EXTEND, 32% and 50% of patients continuing abrocitinib 100 mg once daily in EXTEND achieved EASI-75 by Week 16 and Week 24 (with 4 and 12 additional weeks of treatment), respectively, and 35% and 59% of patients continuing abrocitinib 200 mg once daily achieved EASI-75 response by Week 16 and Week 24, respectively.
Long-term efficacy Among patients who achieved response at Week 12 of a qualifying parent study and entered EXTEND, the majority of patients maintained their response at Week 48 of cumulative abrocitinib treatment for both doses of abrocitinib [60% and 70% for IGA (0 or 1) response, 79% and 87% for EASI-75, and 62% and 83% for PP-NRS4 with 100 mg once daily and 200 mg once daily, respectively].
Health related outcomes Treatment with either dose of abrocitinib as monotherapy resulted in significantly improved patient-reported outcomes at 12 weeks compared with placebo (see Table 7). A significantly larger proportion of the abrocitinib groups had clinically meaningful reductions in Dermatology Life Quality Index (DLQI) total scores (defined as a 4-point improvement) from baseline to Week 12 compared with placebo. abrocitinib groups also had a significantly larger proportion of patients who reported “no effect” of their disease on their quality of life (as measured by a DLQI score of 0 or 1).
Both groups significantly improved patient-reported atopic dermatitis symptoms and sleep disruption as measured by the Patient Oriented Eczema Measure (POEM), Night Time Itch Scale (NTIS), and SCORing Atopic Dermatitis (SCORAD) sleep loss subscale. In addition, anxiety and depression symptoms as measured by the Hospital Anxiety and Depression Scale (HADS) total score were significantly reduced in the abrocitinib groups compared with placebo at 12 weeks.
Table 7. Additional endpoint results with abrocitinib monotherapy at Week 12
MONO-1
| ABR | ABR | ||
|---|---|---|---|
| 200 mg OD N=154 | 100 mg OD N=156 | Placebo N=77 | |
| LSM SCORAD (sleep loss subscale): Baseline median (SD), Change from baseline (95% CI) | 5.9,-3.7 (d) (-4.2, -3.3) | 6.0/-2.9 (c) (-3.4, -2.5) | 6.5 -1.6 (-2.2, -1.0) |
| NTIS > 4-point improvement: % responders | n/a | n/a | n/a |
| DLQI: 0 or 1, % responders ≥ 4 point improvement, % responders | 31.9 (b), 72.6 (c) | 20.2, 67.2 (b) | 12.1, 43.6 |
| LSM DLQI: Baseline mean (SD), Change from baseline (95% CI) | 14.6 (6.8), -9.1 (d) (-10.3, -8.0) | 14.6 (6.5), -7.0 (b) (-8.1, -5.8) | 13.9 (7.3), -4.2 (-5.9, -2.5) |
| CDLQI: ≥ 2.5 point improvement, % responders | 83.9 (a) | 73.3 | 53.3 |
| LSM-CDLQI: Baseline mean (SD), Change from baseline (95% CI) | 13.2 (5.5), -7.5 (a) (-8.9, -6.0) | 11.7 (6.6), -6.4 (-7.9, -5.0) | 13.6 (7.0), -3.9 (-6.1, -1.7) |
| LSM POEM: Baseline mean (SD), Change from baseline (95% CI) | 19.6 (5.9), -10.6 (d) (-11.8, -9.4) | 19.5 (6.5), -6.8 (b) (-8.0, -5.6) | 19.9 (6.1), -3.7 (-5.5, -1.9) |
| LSM HADS (anxiety): Baseline mean (SD), Change from baseline (95% CI) | 5.6 (4.0), -2.1 (b) (-2.5, -1.6) | 5.9 (4.1), -1.6 (-2.0, -1.1) | 6.0 (4.0), -1.0 (-1.7, -0.4) |
| LSM HADS (depression): Baseline mean (SD), Change from baseline (95% CI) | 4.2 (3.7), -1.8 (d) (-2.2, -1.4) | 4.1 (3.7), -1.4 (b) (-1.8, -0.9) | 3.9 (3.5), -0.2 (-0.8, 0.4) |
MONO-2
| ABR | ABR | ||
|---|---|---|---|
| 200 mg OD N=155 | 100 mg OD N=158 | Placebo N=78 | |
| LSM SCORAD (sleep loss subscale): Baseline median (SD), Change from baseline (95% CI) | 6.2, -3.8 (d) (-4.2, -3.4) | 6.2, -3.0 (a) (-3.4, -2.6) | 5.7, -2.1 (-2.7, -1.5) |
| NTIS > 4-point improvement: % responders | 57.0 (d) | 42.7 (d) | 12.7 |
| DLQI: 0 or 1, % responders ≥ 4 point improvement, % responders | 26.6 (c), 78.1 (d) | 20.3 (b), 73.3 (d) | 5.7, 32.3 |
| LSM DLQI: Baseline mean (SD), Change from baseline (95% CI) | 14.8 (6.0), -9.8 (d) (-10.7, -8.8) | 15.4 (7.3), -8.3 (d) (-9.3, -7.3) | 15.0 (7.1), -3.9 (-5.3, -2.4) |
| CDLQI: ≥ 2.5 point improvement, % responders | 93.3 (c) | 56.3 (a) | 12.5 |
| LSM-CDLQI: Baseline mean (SD), Change from baseline (95% CI) | 12.9 (5.7), -9.7 (b) (-12.1, -7.4) | 13.8 (5.8), -4.8 (-7.2, -2.5) | 10.1 (3.8), -2.7 (-6.1, 0.8) |
| LSM POEM: Baseline mean (SD), Change from baseline (95% CI) | 19.7 (5.7), -11.0 (d) (-12.1, -9.8) | 20.9 (5.7), -8.7 (d) (-9.9, -7.5) | 19.2 (5.5), -3.6 (-5.3, -1.9) |
| LSM HADS (anxiety): Baseline mean (SD), Change from baseline (95% CI) | 5.9 (3.9), -1.7 (a) (-2.2, -1.2) | 5.5 (4.2), -1.6 (a) (-2.1, -1.1) | 6.0 (3.7), -0.6 (-1.3, 0.2) |
| LSM HADS (depression): Baseline mean (SD), Change from baseline (95% CI) | 4.0 (3.7), -1.4 (d) (-1.8, -1.0) | 4.1 (4.0), -1.0 (c) (-1.5, -0.6) | 4.4 (3.3), 0.3 (-0.3, 0.9) |
Abbreviations: ABR=abrocitinib; CDLQ =Child Dermatology Life Quality Index; CI=confidence interval; DLQI=Dermatology Life Quality Index; HADS=Hospital Anxiety and Depression Scale; LSM=least squares mean; N=number of patients randomised; n/a= not available; NTIS=Night Time Itch Scale; POEM=Patient Oriented Eczema Measure; OD=once daily; SCORAD=SCORing Atopic Dermatitis.
a. Nominal p < 0.05 versus placebo.
b. Nominal p < 0.01 versus placebo.
c. Nominal p < 0.001 versus placebo.
d. Nominal p < 0.0001 versus placebo.
In COMPARE, a significantly larger proportion of the abrocitinib groups had clinically meaningful reductions in DLQI total scores (defined as a 4 point improvement) from baseline to Week 12 compared with placebo (see Table 8). Abrocitinib groups also had a significantly larger proportion of patients who reported “no effect” of their disease on their quality of life (as measured by a DLQI score of 0 or 1).
Both groups significantly improved patient-reported atopic dermatitis symptoms and sleep disruption as measured by the POEM and SCORAD sleep loss subscale, respectively. In addition, anxiety and depression symptoms as measured by the HADS total score were significantly reduced in the abrocitinib groups compared with placebo at 12 weeks.
Table 8. Additional secondary endpoint results with abrocitinib in combination with medicated topical therapies at Week 12
COMPARE
| ABR | ABR | ||
|---|---|---|---|
| 200 mg OD + Topical N=226 | 100 mg OD + N=238 | Placebo + Topical N=131 | |
| LSM SCORAD (sleep loss subscale): Baseline median (SD), Change from baseline (95% CI) | 6.4, -4.6 (d) (-4.9, -4.3) | 6.1, -3.7 (d) (-4.0, -3.4) | 6.0, -2.4 (-2.8, -2.0) |
| NTIS > 4-point improvement: % responders | 64.3 (d) | 54.0 (c) | 34.4 |
| DLQI: 0 or 1, % responders ≥ 4 point improvement, % responders | 29.7% (d), 86.4% (d) | 21.9% (b), 74.7% (c) | 8.6%, 56.5% |
| LSM DLQI: Baseline mean (SD), Change from baseline (95% CI) | 16.3 (6.6), -11.0 (d) (-11.7, -10.3) | 15.5 (6.4), -8.7 (d) (-9.4, -8.0) | 15.2 (6.9), -6.2 (-7.1, -5.3) |
| LSM POEM: Baseline mean (SD), Change from baseline (95% CI) | 21.5 (5.3), -12.6 (d) (-13.6, -11.7) | 20.9 (5.5), -9.6 (d) (-10.5, -8.6) | 20.4 (6.1), -5.1 (-6.3, -3.9) |
| LSM HADS (anxiety): Baseline mean (SD), Change from baseline (95% CI) | 5.5 (3.8), -1.6 (c) (-2.0, -1.2) | 5.3 (3.9), -1.2 (a) (-1.5, -0.8) | 5.3 (3.9), -0.4 (-0.9, 0.1) |
| LSM HADS (depression): Baseline mean (SD), Change from baseline (95% CI) | 3.9 (3.4), -1.6 (d) (-1.9, -1.2) | 4.0 (3.3), -1.3 (c) (-1.6, -0.9) | 4.1 (3.7), -0.3 (-0.7, 0.2) |
Abbreviations: ABR=abrocitinib; DLQI=Dermatology Life Quality Index; HADS=Hospital Anxiety and Depression Scale; LSM=least squares mean; NTIS=Night Time Itch Scale; POEM=Patient Oriented Eczema Measure; OD=once daily; SCORAD=SCORing Atopic Dermatitis; SD=standard deviation.
a. Nominal p < 0.05 versus placebo.
b. Nominal p < 0.01 versus placebo.
c. Nominal p < 0.001 versus placebo.
d. Nominal p < 0.0001 versus placebo.
Paediatric population
The safety and efficacy of abrocitinib in paediatric patients under 12 years of age have not yet been established.
Patients under 12 years of age will not be able to initiate treatment under the EAMS.
5.2 Pharmacokinetic properties
The pharmacokinetic profile of abrocitinib is characterised by rapid absorption (peak plasma concentrations are reached within 1 hour), and an elimination half-life of about 5 hours. Steady state plasma concentrations of abrocitinib are achieved within 48 hours after once daily administration.
Absorption
Abrocitinib is well-absorbed with over 91% extent of oral absorption and absolute oral bioavailability of approximately 60%. Both Cmax and AUC of abrocitinib increased dose proportionally up to 400 mg. Co-administration of abrocitinib with a high-fat meal had no clinically relevant effect on abrocitinib exposures (AUC and Cmax increased by approximately 26% and 29%, respectively, and Tmax was prolonged by 2 hours). In clinical studies, abrocitinib was administered without regard to food (see section 4.2).
Distribution
After intravenous administration, the volume of distribution of abrocitinib is about 100 L. Approximately 64%, 37% and 29% of circulating abrocitinib and its active metabolites M1 and M2 are bound to plasma proteins, respectively. Abrocitinib and its active metabolites bind predominantly to albumin. Abrocitinib and its active metabolites distribute equally between red blood cells and plasma.
Biotransformation
The metabolism of abrocitinib is mediated by multiple CYP enzymes, CYP2C19 (~53%), CYP2C9 (~30%), CYP3A4 (~11%) and CYP2B6 (~6%). In a human radiolabeled study, abrocitinib was the most prevalent circulating species, with 3 polar mono-hydroxylated metabolites identified as M1 (3-hydroxypropyl), M2 (2-hydroxypropyl), and M4 (pyrrolidinone pyrimidine). Of the 3 metabolites in circulation, M1 and M2 have similar JAK inhibitory profiles as abrocitinib, while M4 was pharmacologically inactive. The pharmacologic activity of abrocitinib is attributable to the unbound exposures of parent molecule (~60%) as well as M1 (~10%) and M2 (~30%) in systemic circulation. The sum of unbound exposures of abrocitinib, M1 and M2, each expressed in molar units and adjusted for relative potencies, is referred to as the abrocitinib active moiety.
Elimination
Abrocitinib is eliminated primarily by metabolic clearance mechanisms, with less than 1% of the dose excreted in urine as unchanged drug. The metabolites of abrocitinib, M1, M2 and M4 are excreted predominantly in urine, and are substrates of OAT3 transporter.
Special populations
Body weight, gender, genotype, race and age Body weight, gender, CYP2C19/2C9 genotype, race, and age did not have a clinically meaningful effect on abrocitinib exposure (see Section 4.2).
Adolescents (≥12 to <18 years) Based on population pharmacokinetic analysis, mean abrocitinib steady-state exposure in adolescent patients is estimated to be approximately 30% lower compared to adults of the same weight, with similar range of exposures in adult and adolescent patients. These differences in mean exposures were not considered clinically significant.
Paediatric (<12 years) The pharmacokinetics of abrocitinib in paediatric patients under 12 years of age have not been established (see section 4.2).
Renal impairment
In a renal impairment study, patients with severe (eGFR <30 mL/min) and moderate (eGFR 30 to <60 mL/min) renal impairment had approximately 191% and 110% increase in active moiety AUCinf, respectively, compared to patients with normal renal function (eGFR ≥ 90 mL/min; see section 4.2). Pharmacokinetics of abrocitinib have not been determined in patients with mild renal impairment, however, based on the results observed in other groups, a clinically significant increase in abrocitinib active moiety is not expected in patients with mild renal impairment (creatinine clearance 60 to < 90 mL/min). The eGFR in individual patients was estimated using Modification of Diet in Renal Disease (MDRD) formula.
Abrocitinib has not been studied in patients with ESRD on renal replacement therapy (see section 4.2). In Phase 3 clinical studies, abrocitinib was not evaluated in patients with atopic dermatitis with baseline creatinine clearance values less than 40 mL/min.
Hepatic impairment
Patients with mild (Child-Pugh A) and moderate (Child-Pugh B) hepatic impairment had approximately 4% decrease and 15% increase in active moiety AUCinf, respectively, compared to patients with normal hepatic function. These changes are not clinically significant, and no dose adjustment is required in patients with mild or moderate hepatic impairment (see section 4.2). In clinical studies, abrocitinib was not evaluated in patients with severe (Child-Pugh C) hepatic impairment, or in patients screened positive for active hepatitis B or hepatitis C.
5.3 Preclinical safety data
Genotoxicity
Abrocitinib is not mutagenic in the bacterial mutagenicity assay (Ames assay). Although abrocitinib is aneugenic in the in vitro TK6 micronucleus assay, abrocitinib is not aneugenic or clastogenic based on the results of the in vivo rat bone marrow micronucleus assay.
Carcinogenicity
No evidence of tumorigenicity was observed in Tg.rasH2 mice administered abrocitinib for 26 weeks at oral doses up to 75 mg/kg/day and 60 mg/kg/day in female and male mice, respectively. In the 104-week oral carcinogenicity study, abrocitinib resulted in statistically higher incidence of benign thymomas in female rats at exposures greater or equal to 2.8 times the unbound human AUC at the maximum recommended human dose (MRHD) of 200 mg. No evidence of abrocitinib-related tumorigenicity was observed following oral abrocitinib administration in female rats at exposures equal to 0.6 times the unbound human AUC at the MRHD of 200 mg or in male rats at exposures equal to 14 times the unbound human AUC at the MRHD of 200 mg.
Reproductive and developmental toxicity
Abrocitinib had no effects on male fertility or spermatogenesis at doses up to 70 mg/kg/day at exposures equal to 26 times the unbound human AUC at the MRHD of 200 mg. Abrocitinib resulted in effects on female fertility (lower fertility index, corpora lutea, and implantation sites) at exposures equal to 29 times the unbound human AUC at the MRHD of 200 mg and higher post-implantation loss in rats at exposures greater than or equal to 11 times the unbound human AUC at the MRHD of 200 mg. The effects on female fertility in rats reversed 1 month after cessation of abrocitinib administration. No effects on female fertility were noted at exposures equal to 2 times the unbound human AUC at the MRHD of 200 mg.
No fetal malformations were observed in embryo-fetal development studies in rats or rabbits. In an embryo-fetal development study in pregnant rabbits, oral administration of abrocitinib during gestation days 7 to 19 had no effects on embryo-fetal survival or fetal morphological development at exposures equal to 4 times the unbound human AUC at the MRHD of 200 mg. Abrocitinib resulted in an increase incidence of delayed ossification of the forelimb phalanges at exposures equal to 4 times the unbound human AUC at the MRHD of 200 mg.
In an embryo-fetal development study in pregnant rats, oral administration of abrocitinib during gestation days 6 to 17 resulted in increased embryo-fetal lethality at exposures equal to 17 times the unbound human AUC at the MRHD of 200 mg. No embryo-fetal lethality was observed in pregnant rats orally dosed with abrocitinib during organogenesis at exposures equal to 11 times the unbound human AUC at the MRHD of 200 mg. Abrocitinib resulted in increased instances of skeletal variations of increased incidences of short 13th ribs at exposures greater than or equal to 11 times the unbound human AUC at the MRHD of 200 mg and reduced ventral processes, thickened ribs, and unossified metatarsals were observed at exposures equal to 17 times the unbound human AUC at the MRHD of 200 mg. No skeletal variations were noted in rats at exposures equal to 2.4 times the unbound human AUC at the MRHD of 200 mg.
In a rat pre- and postnatal development study in pregnant rats, oral administration of abrocitinib during gestation day 6 through lactation day 21 resulted in dystocia with prolonged parturition and lower offspring body weights at exposures greater than or equal to 11 times the unbound human AUC at the MRHD of 200 mg and lower postnatal survival at exposures equal to 17 times the unbound human AUC at the MRHD of 200 mg. No maternal or developmental toxicity was observed in either dams or offspring at exposures equal to 2.4 times the unbound human AUC at the MRHD of 200 mg.
A summary table of preclinical safety studies, findings, and human safety margins is presented in Table 9.
Table 9. Preclinical safety studies findings and human safety margins
| Preclinical Study Type | Preclinical Safety Finding | Human Safety Margin (a) |
|---|---|---|
| In vivo rat bone marrow micronucleus assay | No aneugenicity or clastogenicity | Up to 121x |
| 6-month Tg mouse carcinogenicity | No evidence of tumorigenicity | Up to 0.9x |
| 104-week rat carcinogenicity | Benign thymoma in females | ≥2.8x |
| 104-week rat carcinogenicity | No evidence of tumorigenicity in females | 0.6x |
| 104-week rat carcinogenicity | No evidence of tumorigenicity in males | 14x |
| Male fertility in rats | No effects on fertility or spermatogenesis | 26x |
| Female fertility in rats | Lower fertility index, corpora lutea, and implantation sites | 29x |
| Female fertility in rats | Higher postimplantation loss | ≥11x |
| Female fertility in rats | No effects on fertility | 2x |
| Embryo-fetal development in rats | No fetal malformations; increased embryo-fetal lethality, higher postimplantation loss; reduced ventral processes, thickened ribs, and unossified metatarsals | 17x |
| Embryo-fetal development in rats | No embryo-fetal lethality | 11x |
| Embryo-fetal development in rats | Increased instances of skeletal variations of short 13th ribs | ≥11x |
| Embryo-fetal development in rats | No skeletal variations | 2.4x |
| Embryo-fetal development in rabbits | Increased incidence of delayed ossification of the forelimb phalanges | 4x |
| Embryo-fetal development in rabbits | No fetal malformations, effects on embryo-fetal survival or fetal morphological development | 4x |
| Pre- and postnatal development in rats | Lower postnatal survival | 17x |
| Pre- and postnatal development in rats | Dystocia with prolonged parturition and lower offspring body weights | ≥11x |
| Pre- and postnatal development in rats | No maternal or developmental toxicity | ≥2.4x |
a. Unbound exposure margins were calculated using animal exposures (unbound values) relative to the MRHD of 200 mg.
6. Pharmaceutical particulars
6.1 List of excipients
Tablet core
- Microcrystalline cellulose
- Dibasic calcium phosphate anhydrous
- Sodium starch glycolate
- Magnesium stearate
Film coat
- Hypromellose (E464)
- Titanium dioxide (E171)
- Lactose monohydrate
- Macrogol/PEG
- Triacetin (E1518)
6.2 Incompatibilities
Not applicable.
6.3 Shelf life
33 months.
6.4 Special precautions for storage
Store at 15 °C – 30 °C.
6.5 Nature and contents of container
High-density polyethylene (HDPE) bottle and closure. Each pack contains 98 film-coated tablets.
6.6 Special precautions for disposal and other handling
Any unused medicinal product or waste material should be disposed of in accordance with local requirements.
7. Scientific opinion holder
Pfizer Limited
Ramsgate Road
Sandwich, Kent
CT13 9NJ
United Kingdom
8. EAMS number
00057/0006
9. Date of scientific opinion
Date of first authorisation: 28 January 2021
Additional information
Each prescribing clinician involved in the patient’s treatment must register and agree to the terms of adverse event reporting. They will have to complete a short Initial Drug Supply and electronic Case Report Form.
An Informed Consent/Assent Form will be provided by Pfizer to be completed with the patient.
A Letter of Agreement will be signed by the prescribing clinician and a legal representative from the trust.
Pfizer will arrange training (including adverse event training) and provision of the programme materials.
A Drug Re-supply and electronic Case Report Form will be accessible online to order further drug supplies. The prescribing clinician will be required to complete the Drug Re-supply and Case Report Form every three months to order the prescription of abrocitinib for their patient. The order should be placed at least two weeks before the next planned prescription is due.
The prescribing clinician is requested to inform Pfizer if a patient discontinues treatment by completing the electronic Case Report Form with the date of the patient’s last treatment and dose.
Contact information
To initiate the registration process for EAMS you are required to obtain an Informed Consent/Assent Form and Initial Drug Supply and Case Report Form from Pfizer by accessing the Pfizer EAMS website. The step by step details on access to abrocitinib, training on adverse events reporting and all forms will be accessible through this site.
For NHS England:
The prescribing clinician should complete the Informed Consent/Assent Form with the patient and submit a NHS EAMS Application form to england.eams@nhs.net to register the patient with NHS England. The NHS application form itself can be requested from the same dedicated NHS EAMS email address above.
Following patient registration with NHS England, the clinician may proceed to request drug supply from Pfizer (using the unique approval number provided by NHS England) for that patient by completing the Initial Drug Supply and Case Report Form with additional eligibility information and submitting this through the Pfizer EAMS website. A copy of the NHS approval email for the patient must also be sent with the form.
For NHS Wales, Scotland and Northern Ireland:
The prescribing clinician should complete the Informed Consent/Assent Form with the patient and may then proceed to request drug supply from Pfizer for that patient by completing the Initial Drug Supply and Case Report Form with additional eligibility information and submitting this through the Pfizer EAMS website. Pfizer will assign a unique patient EAMS number to be used in any future communications.
Additional contact: Pfizer Medical Information on +44 (0) 1304 616161 or medical.information@pfizer.com