Nivolumab: Treatment protocol: Information for healthcare professionals
Published 1 February 2021

© Crown copyright 2021
This publication is licensed under the terms of the Open Government Licence v3.0 except where otherwise stated. To view this licence, visit nationalarchives.gov.uk/doc/open-government-licence/version/3 or write to the Information Policy Team, The National Archives, Kew, London TW9 4DU, or email: psi@nationalarchives.gov.uk.
Where we have identified any third party copyright information you will need to obtain permission from the copyright holders concerned.
This publication is available at https://www.gov.uk/government/publications/nivolumab-with-ipilimumab-in-the-treatment-of-malignant-pleural-mesothelioma/nivolumab-treatment-protocol-information-for-healthcare-professionals
1. Introduction
The aim of the Early Access to Medicines Scheme (EAMS) is to provide earlier availability of promising new unlicensed medicines and medicines used outside their licence, to UK patients that have a high unmet clinical need. The medicinal products included in the scheme are those that are intended to treat, diagnose or prevent seriously debilitating or life-threatening conditions where there are no adequate treatment options. More information about the scheme can be found here: http://www.mhra.gov.uk/Howweregulate/Innovation/EarlyaccesstomedicinesschemeEAMS/index.htm
This information is intended for healthcare professionals and is provided by the pharmaceutical company that manufactures the EAMS medicine. This medicine does not yet have a licence (marketing authorisation) in this indication and is to be used in combination with (an)other medicine(s) prescribed outside the licence. The information is provided to assist physicians in prescribing medicines used outside the licence. Guidance on prescribing unlicensed medicines can be found on the GMC webpage: https://www.gmc-uk.org/guidance/ethical_guidance/14327.asp
The scientific opinion is based on assessment of the information supplied to the MHRA on the benefits and risks of the combination therapy in this new promising indication. As such, this is a scientific opinion and should not be regarded as an indication licensed by the MHRA or a future commitment by the MHRA to license such an indication, nor should it be regarded as an authorisation to sell or supply a medicine for such an indication. A positive scientific opinion is not a recommendation for use of the medicine and should not be interpreted as such. Under EAMS the risk and legal responsibility for prescribing a ‘special’ remains with the physician, and the opinion and EAMs documentation published by the MHRA are intended only to inform physicians’ decision making and not to recommend use. An EAMS scientific opinion does not affect the civil liability of the manufacturer or any physician in relation to the product.
Healthcare professionals should also refer to the summary information on the pharmacovigilance system which is provided in the document ‘Early Access to Medicines Scheme – Treatment protocol – Information on the pharmacovigilance system’.
Scientific opinion period: The MHRA will withdraw the EAMS positive scientific opinion when a marketing authorisation (drug licence) is issued for the EAMS product covering the EAMS indication, or if following scientific assessment, the EAMS criteria are considered to be no longer met.
Treatment protocol update(s): In case of substantial new efficacy or safety data, the treatment protocol may need to be updated. For other updates of the safety information, please refer to the product information of the combination products on the electronic Medicines Compendium (eMC) website: https://www.medicines.org.uk/emc
Contact information regarding queries on using this EAMS medicine can be found at the end of this document.
2. Information for the healthcare professionals:
2.1 1. NAME OF THE MEDICINAL PRODUCT
Nivolumab 10 mg/mL concentrate for solution for infusion.
2.2 2. QUALITATIVE AND QUANTITATIVE COMPOSITION
Nivolumab Each mL of concentrate contains 10 mg of nivolumab. One vial of 10 mL contains 100 mg of nivolumab.
Nivolumab is produced in Chinese hamster ovary cells by recombinant DNA technology.
Ipilimumab Each ml of concentrate contains 5 mg ipilimumab. One 40 ml vial contains 200 mg of ipilimumab.
Ipilimumab is a fully human anti CTLA 4 monoclonal antibody (IgG1κ) produced in Chinese hamster ovary cells by recombinant DNA technology.
Excipient with known effect
Nivolumab: Each mL of concentrate contains 0.1 mmol (or 2.5 mg) sodium. Ipilimumab: Each ml of concentrate contains 0.1 mmol sodium, which is 2.30 mg sodium.
For the full list of excipients of nivolumab, see section 6.1. For the full list of excipients of ipilimumab, please refer to section 6.1 of the Yervoy (ipilimumab) Summary of Product Characteristics (SmPC).
2.3 3. PHARMACEUTICAL FORM
Nivolumab: Concentrate for solution for infusion (sterile concentrate). Clear to opalescent, colourless to pale yellow liquid that may contain few light particles. The solution has a pH of approximately 6.0 and an osmolality of approximately 340 mOsm/kg.
Ipilimumab: Concentrate for solution for infusion (sterile concentrate). Clear to slightly opalescent, colourless to pale yellow liquid that may contain light (few) particulates and has a pH of 7.0 and an osmolarity of 260-300 mOsm/kg.
2.4 4. CLINICAL PARTICULARS
4.1 EAMS therapeutic indication
Nivolumab in combination with ipilimumab is indicated for the first-line treatment of adult patients with unresectable malignant pleural mesothelioma.
Healthcare professionals are advised to consult the Summaries of Product Characteristics (SmPCs) for Opdivo (nivolumab) and ipilimumab for further information on administration, safety aspects and pharmaceutical particulars via the links below:
- nivolumab: https://www.medicines.org.uk/emc/product/6888
- ipilimumab: https://www.medicines.org.uk/emc/product/4683
4.2 Posology and method of administration
Treatment must be initiated and supervised by physicians experienced in the treatment of cancer.
Posology
The recommended dose is 360 mg nivolumab administered intravenously over 30 minutes every 3 weeks in combination with 1 mg/kg ipilimumab administered intravenously over 30 minutes every 6 weeks. Treatment is continued for up to 24 months in patients without disease progression.
Duration of treatment Treatment with nivolumab, either as monotherapy or in combination with ipilimumab, should be continued as long as clinical benefit is observed or until treatment is no longer tolerated by the patient.
Atypical responses (i.e., an initial transient increase in tumour size or small new lesions within the first few months followed by tumour shrinkage) have been observed. It is recommended to continue treatment with nivolumab or nivolumab in combination with ipilimumab for clinically stable patients with initial evidence of disease progression until disease progression is confirmed.
Dose escalation or reduction is not recommended. Dosing delay or discontinuation may be required based on individual safety and tolerability. Guidelines for permanent discontinuation or withholding of doses are described in Table 1. Detailed guidelines for the management of immune-related adverse reactions are described in section 4.4.
Table 1: Recommended treatment modifications for nivolumab in combination with ipilimumab
| Immune-related adverse reaction | Severity | Treatment modification |
|---|---|---|
| Immune-related pneumonitis | Grade 2 pneumonitis | Withhold dose(s) until symptoms resolve, radiographic abnormalities improve, and management with corticosteroids is complete |
| Immune-related pneumonitis | Grade 3 or 4 pneumonitis | Permanently discontinue treatment |
| Immune-related colitis | Grade 2 diarrhoea or colitis | Withhold dose(s) until symptoms resolve and management with corticosteroids, if needed, is complete |
| Immune-related colitis | Grade 3 diarrhoea or colitis | Permanently discontinue treatment |
| Immune-related colitis | Grade 4 diarrhoea or colitis | Permanently discontinue treatment |
| Immune-related hepatitis | Grade 2 elevation in aspartate aminotransferase (AST), alanine aminotransferase (ALT), or total bilirubin | Withhold dose(s) until laboratory values return to baseline and management with corticosteroids, if needed, is complete |
| Immune-related hepatitis | Grade 3 or 4 elevation in AST, ALT, or total bilirubin | Permanently discontinue treatment |
| Immune-related nephritis and renal dysfunction | Grade 2 or 3 creatinine elevation | Withhold dose(s) until creatinine returns to baseline and management with corticosteroids is complete |
| Immune-related nephritis and renal dysfunction | Grade 4 creatinine elevation | Permanently discontinue treatment |
| Immune-related endocrinopathies | Symptomatic Grade 2 or 3 hypothyroidism, hyperthyroidism, hypophysitis, Grade 2 adrenal insufficiency, Grade 3 diabetes | Withhold dose(s) until symptoms resolve and management with corticosteroids (if needed for symptoms of acute inflammation) is complete. Treatment should be continued in the presence of hormone replacement therapya as long as no symptoms are present |
| Immune-related endocrinopathies | Grade 4 hypothyroidism, Grade 4 hyperthyroidism , Grade 4 hypophysitis, Grade 3 or 4 adrenal insufficiency, Grade 4 diabetes | Permanently discontinue treatment |
| Immune-related skin adverse reactions | Grade 3 rash | Withhold dose(s) until symptoms resolve and management with corticosteroids is complete |
| Immune-related skin adverse reactions | Grade 4 rash | Permanently discontinue treatment |
| Immune-related skin adverse reactions | Stevens-Johnson syndrome (SJS) or toxic epidermal necrolysis (TEN) | Permanently discontinue treatment (see section 4.4) |
| Immune-related myocarditis | Grade 2 myocarditis | Withhold dose(s) until symptoms resolve and management with corticosteroids is complete (b) |
| Immune-related myocarditis | Grade 3 or 4 myocarditis | Permanently discontinue treatment |
| Other immune-related adverse reactions | Grade 3 (first occurrence) | Withhold dose(s) |
| Other immune-related adverse reactions | Grade 4 or recurrent Grade 3 ; persistent Grade 2 or 3 despite treatment modification; inability to reduce corticosteroid dose to 10 mg prednisone or equivalent per day | Permanently discontinue treatment |
Note: Toxicity grades are in accordance with National Cancer Institute Common Terminology Criteria for Adverse Events Version 4.0 (NCI-CTCAE v4).
a Recommendation for the use of hormone replacement therapy is provided in section 4.4.
b The safety of re-initiating nivolumab in combination with ipilimumab therapy in patients previously experiencing immune-related myocarditis is not known
Nivolumab in combination with ipilimumab should be permanently discontinued for
-
Grade 4 or recurrent Grade 3 adverse reactions;
-
Persistent Grade 2 or 3 adverse reactions despite management.
Patients treated with nivolumab in combination with ipilimumab must be given the patient alert card and be informed about the risks of nivolumab and ipilimumab (see also EAMS – Treatment Protocol – Information for Patients and the ipilimumab patient leaflet).
When nivolumab is administered in combination with ipilimumab, if either agent is withheld, the other agent should also be withheld. If dosing is resumed after a delay, either the combination treatment or nivolumab monotherapy could be resumed based on the evaluation of the individual patient.
If treatment is continued with nivolumab only, please consult the Opdivo SmPC for guidelines on the management of immune-related adverse reactions, administration, safety aspects and pharmaceutical particulars.
Special populations
Paediatric population The safety and efficacy of nivolumab in children below 18 years of age have not been established. No data are available.
Elderly No dose adjustment is required for elderly patients (≥ 65 years) (see section 5.2).
Renal impairment Based on the population pharmacokinetic (PK) results, no dose adjustment is required in patients with mild or moderate renal impairment (see section 5.2). Data from patients with severe renal impairment are too limited to draw conclusions on this population.
Hepatic impairment Based on the population PK results, no dose adjustment is required in patients with mild hepatic impairment (see section 5.2). Data from patients with moderate or severe hepatic impairment are too limited to draw conclusions on these populations. Nivolumab must be administered with caution in patients with moderate (total bilirubin > 1.5 × to 3 × the upper limit of normal [ULN] and any AST) or severe (total bilirubin > 3 × ULN and any AST) hepatic impairment.
Method of administration
Nivolumab and ipilimumab are for intravenous use only. Each product is to be administered as an intravenous infusion over a period of 30 minutes. The infusion must be administered through a sterile, non-pyrogenic, low protein binding in-line filter with a pore size of 0.2-1.2 μm.
Nivolumab and ipilimumab must not be administered as an intravenous push or bolus injection.
The total dose of nivolumab required can be infused directly as a 10 mg/mL solution or can be diluted with sodium chloride 9 mg/mL (0.9%) solution for injection or glucose 50 mg/mL (5%) solution for injection (see section 6.6).
When administered in combination with ipilimumab, nivolumab should be given first followed by ipilimumab on the same day. Use separate infusion bags and filters for each infusion.
For instructions on the preparation and handling of nivolumab before administration, see section 6.6. Healthcare professionals are advised to consult the SmPC for ipilimumab for information on method of administration for this product.
2.5 4.3 Contraindications
Hypersensitivity to the active substance or to any of the excipients listed in section 6.1.
2.6 4.4 Special warnings and precautions for use
Traceability In order to improve the traceability of biological medicinal products, the EAMS number and batch numbers of the administered product(s) should be clearly recorded in the patient file.
Immune-related adverse reactions When nivolumab is administered in combination with ipilimumab, refer to the Summary of Product Characteristics for ipilimumab prior to initiation of treatment. Immune-related adverse reactions have occurred at higher frequencies when nivolumab was administered in combination with ipilimumab compared with nivolumab as monotherapy. Most immune-related adverse reactions improved or resolved with appropriate management, including initiation of corticosteroids and treatment modifications (see section 4.2).
Cardiac and pulmonary adverse reactions including pulmonary embolism have also been reported with combination therapy. Patients should be monitored for cardiac and pulmonary adverse reactions continuously, as well as for clinical signs, symptoms, and laboratory abnormalities indicative of electrolyte disturbances and dehydration prior to and periodically during treatment. Nivolumab in combination with ipilimumab should be discontinued for life-threatening or recurrent severe cardiac and pulmonary adverse reactions (see section 4.2).
Patients should be monitored continuously (at least up to 5 months after the last dose) as an adverse reaction with nivolumab in combination with ipilimumab may occur at any time during or after discontinuation of therapy.
For suspected immune-related adverse reactions, adequate evaluation should be performed to confirm aetiology or exclude other causes. Based on the severity of the adverse reaction, nivolumab in combination with ipilimumab should be withheld and corticosteroids administered. If immunosuppression with corticosteroids is used to treat an adverse reaction, a taper of at least 1 month duration should be initiated upon improvement. Rapid tapering may lead to worsening or recurrence of the adverse reaction. Non-corticosteroid immunosuppressive therapy should be added if there is worsening or no improvement despite corticosteroid use.
Nivolumab in combination with ipilimumab should not be resumed while the patient is receiving immunosuppressive doses of corticosteroids or other immunosuppressive therapy. Prophylactic antibiotics should be used to prevent opportunistic infections in patients receiving immunosuppressive therapy.
Nivolumab in combination with ipilimumab must be permanently discontinued for any severe immune-related adverse reaction that recurs and for any life-threatening immune-related adverse reaction.
If treatment is continued with nivolumab only, please consult the Opdivo SmPC for guidelines on the management of immune-related adverse reactions, administration, safety aspects and pharmaceutical particulars.
Immune-related pneumonitis Severe pneumonitis or interstitial lung disease, including fatal cases, has been observed with nivolumab in combination with ipilimumab (see section 4.8). Patients should be monitored for signs and symptoms of pneumonitis such as radiographic changes (e.g., focal ground glass opacities, patchy filtrates), dyspnoea, and hypoxia. Infectious and disease-related aetiologies should be ruled out.
For Grade 3 or 4 pneumonitis, nivolumab in combination with ipilimumab must be permanently discontinued, and corticosteroids should be initiated at a dose of 2 to 4 mg/kg/day methylprednisolone equivalents.
For Grade 2 (symptomatic) pneumonitis, nivolumab in combination with ipilimumab should be withheld and corticosteroids initiated at a dose of 1 mg/kg/day methylprednisolone equivalents. Upon improvement, nivolumab in combination with ipilimumab may be resumed after corticosteroid taper. If worsening or no improvement occurs despite initiation of corticosteroids, corticosteroid dose should be increased to 2 to 4 mg/kg/day methylprednisolone equivalents and nivolumab in combination with ipilimumab must be permanently discontinued.
Immune-related colitis Severe diarrhoea or colitis has been observed with nivolumab in combination with ipilimumab (see section 4.8). Patients should be monitored for diarrhoea and additional symptoms of colitis, such as abdominal pain and mucus or blood in stool. Cytomegalovirus (CMV) infection/reactivation has been reported in patients with corticosteroid-refractory immune-related colitis. Infectious and other aetiologies of diarrhoea should be ruled out, therefore appropriate laboratory tests and additional examinations must be performed. If diagnosis of corticosteroid-refractory immune-related colitis is confirmed addition of an alternative immunosuppressive agent to the corticosteroid therapy, or replacement of the corticosteroid therapy, should be considered.
For Grade 4 diarrhoea or colitis, nivolumab in combination with ipilimumab must be permanently discontinued, and corticosteroids should be initiated at a dose of 1 to 2 mg/kg/day methylprednisolone equivalents.
Grade 3 diarrhoea or colitis observed with nivolumab in combination with ipilimumab requires permanent discontinuation of treatment and initiation of corticosteroids at a dose of 1 to 2 mg/kg/day methylprednisolone equivalents.
For Grade 2 diarrhoea or colitis, nivolumab in combination with ipilimumab should be withheld. Persistent diarrhoea or colitis should be managed with corticosteroids at a dose of 0.5 to 1 mg/kg/day methylprednisolone equivalents. Upon improvement, nivolumab in combination with ipilimumab may be resumed after corticosteroid taper, if needed. If worsening or no improvement occurs despite initiation of corticosteroids, corticosteroid dose should be increased to 1 to 2 mg/kg/day methylprednisolone equivalents and nivolumab in combination with ipilimumab must be permanently discontinued.
Immune-related hepatitis Severe hepatitis has been observed with nivolumab in combination with ipilimumab (see section 4.8). Patients should be monitored for signs and symptoms of hepatitis such as transaminase and total bilirubin elevations. Infectious and disease-related aetiologies should be ruled out.
For Grade 3 or 4 transaminase or total bilirubin elevation, nivolumab in combination with ipilimumab must be permanently discontinued, and corticosteroids should be initiated at a dose of 1 to 2 mg/kg/day methylprednisolone equivalents.
For Grade 2 transaminase or total bilirubin elevation nivolumab in combination with ipilimumab should be withheld. Persistent elevations in these laboratory values should be managed with corticosteroids at a dose of 0.5 to 1 mg/kg/day methylprednisolone equivalents. Upon improvement, nivolumab in combination with ipilimumab may be resumed after corticosteroid taper, if needed. If worsening or no improvement occurs despite initiation of corticosteroids, corticosteroid dose should be increased to 1 to 2 mg/kg/day methylprednisolone equivalents and nivolumab in combination with ipilimumab must be permanently discontinued.
Immune-related nephritis and renal dysfunction Severe nephritis and renal dysfunction have been observed with nivolumab in combination with ipilimumab (see section 4.8). Patients should be monitored for signs and symptoms of nephritis or renal dysfunction. Most patients present with asymptomatic increases in serum creatinine. Disease-related aetiologies should be ruled out.
For Grade 4 serum creatinine elevation, nivolumab in combination with ipilimumab must be permanently discontinued, and corticosteroids should be initiated at a dose of 1 to 2 mg/kg/day methylprednisolone equivalents.
For Grade 2 or 3 serum creatinine elevation, nivolumab in combination with ipilimumab should be withheld, and corticosteroids should be initiated at a dose of 0.5 to 1 mg/kg/day methylprednisolone equivalents. Upon improvement, nivolumab in combination with ipilimumab may be resumed after corticosteroid taper. If worsening or no improvement occurs despite initiation of corticosteroids, corticosteroid dose should be increased to 1 to 2 mg/kg/day methylprednisolone equivalents, and nivolumab in combination with ipilimumab must be permanently discontinued.
Immune-related endocrinopathies Severe endocrinopathies, including hypothyroidism, hyperthyroidism, adrenal insufficiency (including secondary adrenocortical insufficiency), hypophysitis (including hypopituitarism), diabetes mellitus, and diabetic ketoacidosis have been observed with nivolumab in combination with ipilimumab (see section 4.8).
Patients should be monitored for clinical signs and symptoms of endocrinopathies and for hyperglycaemia and changes in thyroid function (at the start of treatment, periodically during treatment, and as indicated based on clinical evaluation). Patients may present with fatigue, headache, mental status changes, abdominal pain, unusual bowel habits, and hypotension, or nonspecific symptoms which may resemble other causes such as brain metastasis or underlying disease. Unless an alternate aetiology has been identified, signs or symptoms of endocrinopathies should be considered immune-related.
For symptomatic hypothyroidism, nivolumab in combination with ipilimumab should be withheld, and thyroid hormone replacement should be initiated as needed. For symptomatic hyperthyroidism, nivolumab in combination with ipilimumab should be withheld and antithyroid medication should be initiated as needed. Corticosteroids at a dose of 1 to 2 mg/kg/day methylprednisolone equivalents should also be considered if acute inflammation of the thyroid is suspected. Upon improvement, nivolumab in combination with ipilimumab may be resumed after corticosteroid taper, if needed. Monitoring of thyroid function should continue to ensure appropriate hormone replacement is utilised. Nivolumab in combination with ipilimumab must be permanently discontinued for life-threatening hyperthyroidism or hypothyroidism.
For symptomatic Grade 2 adrenal insufficiency, nivolumab in combination with ipilimumab should be withheld, and physiologic corticosteroid replacement should be initiated as needed. Nivolumab in combination with ipilimumab must be permanently discontinued for severe (Grade 3) or life-threatening (Grade 4) adrenal insufficiency. Monitoring of adrenal function and hormone levels should continue to ensure appropriate corticosteroid replacement is utilised.
For symptomatic Grade 2 or 3 hypophysitis, nivolumab in combination with ipilimumab should be withheld, and hormone replacement should be initiated as needed. Corticosteroids at a dose of 1 to 2 mg/kg/day methylprednisolone equivalents should also be considered if acute inflammation of the pituitary gland is suspected. Upon improvement, nivolumab in combination with ipilimumab may be resumed after corticosteroid taper, if needed. Nivolumab in combination with ipilimumab must be permanently discontinued for life-threatening (Grade 4) hypophysitis. Monitoring of pituitary function and hormone levels should continue to ensure appropriate hormone replacement is utilised.
For symptomatic diabetes, nivolumab in combination with ipilimumab should be withheld, and insulin replacement should be initiated as needed. Monitoring of blood sugar should continue to ensure appropriate insulin replacement is utilised. Nivolumab in combination with ipilimumab must be permanently discontinued for life-threatening diabetes.
Immune-related skin adverse reactions Severe rash has been observed with nivolumab in combination with ipilimumab (see section 4.8). Nivolumab in combination with ipilimumab should be withheld for Grade 3 rash and discontinued for Grade 4 rash. Severe rash should be managed with high-dose corticosteroid at a dose of 1 to 2 mg/kg/day methylprednisolone equivalents.
Rare cases of SJS and TEN some of them with fatal outcome have been observed. If symptoms or signs of SJS or TEN appear, treatment with nivolumab in combination with ipilimumab should be discontinued and the patient referred to a specialised unit for assessment and treatment. If the patient has developed SJS or TEN with the use of nivolumab in combination with ipilimumab, permanent discontinuation of treatment is recommended (see section 4.2).
Caution should be used when considering the use of nivolumab in a patient who has previously experienced a severe or life-threatening skin adverse reaction on prior treatment with other immune-stimulatory anticancer agents.
Other immune-related adverse reactions The following immune-related adverse reactions were reported in less than 1% of patients treated with nivolumab in combination with ipilimumab in clinical trials across doses and tumour types: pancreatitis, uveitis, demyelination, autoimmune neuropathy (including facial and abducens nerve paresis), Guillain-Barré syndrome, myasthenia gravis, myasthenic syndrome, aseptic meningitis, encephalitis, gastritis, sarcoidosis, duodenitis, myositis, myocarditis, and rhabdomyolysis. Cases of Vogt-Koyanagi-Harada syndrome and hypoparathyroidism have been reported post-marketing (see section 4.8 of this document and section 4.8 in the nivolumab SmPC).
For suspected immune-related adverse reactions, adequate evaluation should be performed to confirm aetiology or exclude other causes. Based on the severity of the adverse reaction, nivolumab in combination with ipilimumab should be withheld and corticosteroids administered.
Upon improvement, nivolumab in combination with ipilimumab may be resumed after corticosteroid taper. Nivolumab in combination with ipilimumab must be permanently discontinued for any severe immune-related adverse reaction that recurs and for any life-threatening immune-related adverse reaction.
Cases of myotoxicity (myositis, myocarditis, and rhabdomyolysis), some with fatal outcome, have been reported with nivolumab in combination with ipilimumab. If a patient develops signs and symptoms of myotoxicity, close monitoring should be implemented, and the patient referred to a specialist for assessment and treatment without delay. Based on the severity of myotoxicity, nivolumab in combination with ipilimumab should be withheld or discontinued (see section 4.2), and appropriate treatment instituted.
The diagnosis of myocarditis requires a high index of suspicion. Patients with cardiac or cardio- pulmonary symptoms should be assessed for potential myocarditis. If myocarditis is suspected, prompt initiation of a high dose of steroids (prednisone 1 to 2 mg/kg/day or methylprednisolone 1 to 2 mg/kg/day) and prompt cardiology consultation with diagnostic workup according to current clinical guidelines should be initiated. Once a diagnosis of myocarditis is established, nivolumab in combination with ipilimumab should be withheld or permanently discontinued (see section 4.2).
Solid organ transplant rejection has been reported in the post-marketing setting in patients treated with PD-1 inhibitors. Treatment with nivolumab may increase the risk of rejection in solid organ transplant recipients. The benefit of treatment with nivolumab versus the risk of possible organ rejection should be considered in these patients.
Haemophagocytic lymphohistiocytosis (HLH) has been observed with nivolumab in combination with ipilimumab. Caution should be taken when nivolumab is administered in combination with ipilimumab. If HLH is confirmed, administration of nivolumab in combination with ipilimumab should be discontinued and treatment for HLH initiated.
Infusion reactions Severe infusion reactions have been reported in clinical trials of nivolumab in combination with ipilimumab (see section 4.8). In case of a severe or life-threatening infusion reaction, the nivolumab in combination with ipilimumab infusion must be discontinued and appropriate medical therapy administered. Patients with mild or moderate infusion reaction may receive nivolumab in combination with ipilimumab with close monitoring and use of premedication according to local treatment guidelines for prophylaxis of infusion reactions.
Disease-specific precautions
Malignant pleural mesothelioma (MPM) Patients with primitive peritoneal, pericardial, testis, or tunica vaginalis mesothelioma, interstitial lung disease, active autoimmune disease, medical conditions requiring systemic immunosuppression,and brain metastasis (unless surgically resected or treated with stereotaxic radiotherapy and no evolution within 3 months prior to inclusion in the study) were excluded from the pivotal trial in first-line treatment of MPM (see sections 4.5 and 5.1). In the absence of data, nivolumab in combination with ipilimumab should be used with caution in these populations after careful consideration of the potential benefit/risk on an individual basis.
Complications of allogeneic haematopoietic stem cell transplant (HSCT) in classical Hodgkin lymphoma Preliminary results from the follow-up of patients with cHL undergoing allogeneic HSCT after previous exposure to nivolumab showed a higher than expected number of cases of acute graft-versus-host disease (GVHD) and transplant related mortality (TRM). Until further data become available, careful consideration to the potential benefits of HSCT and the possible increased risk of transplant related complications should be made case-by-case (see section 4.8).
In patients treated with nivolumab after allogeneic HSCT, rapid-onset and severe GVHD, some with fatal outcome, have been reported in the post-marketing setting. Treatment with nivolumab may increase the risk of severe GVHD and death in patients who have had prior allogeneic HSCT, mainly in those with prior history of GVHD. The benefit of treatment with nivolumab versus the possible risk should be considered in these patients (see section 4.8).
For information on other disease-specific precautions, please refer to the SmPCs for nivolumab and ipilimumab.
Patients on controlled sodium diet
Nivolumab: Each mL of this medicinal product contains 0.1 mmol (or 2.5 mg) sodium. This medicinal product contains 25 mg sodium per 10 ml vial, which is equivalent to 1.25% of the WHO recommended maximum daily intake of 2 g sodium for an adult.
Ipilimumab: This medicinal product contains 92 mg sodium per 40 ml vial, equivalent to 4.60% of the WHO recommended maximum daily intake of 2 g sodium for an adult. To be taken into consideration when treating patients on a controlled sodium diet.
Patient Alert Card The prescriber must discuss the risks of nivolumab and ipilimumab combination therapy with the patient and provide a Patient Alert Card to each patient prior to starting treatment.
2.7 4.5 Interaction with other medicinal products and other forms of interaction
Nivolumab is a human monoclonal antibody, as such pharmacokinetic interaction studies have not been conducted. As monoclonal antibodies are not metabolised by cytochrome P450 (CYP) enzymes or other drug metabolising enzymes, inhibition or induction of these enzymes by co-administered medicinal products is not anticipated to affect the pharmacokinetics of nivolumab.
Other forms of interaction
Systemic immunosuppression The use of systemic corticosteroids and other immunosuppressants at baseline, before starting nivolumab, should be avoided because of their potential interference with the pharmacodynamic activity. However, systemic corticosteroids and other immunosuppressants can be used after starting nivolumab to treat immune-related adverse reactions. The preliminary results show that systemic immunosuppression after starting nivolumab treatment does not appear to preclude the response on nivolumab.
2.8 4.6 Fertility, pregnancy and lactation
Pregnancy There are no data from the use of nivolumab in pregnant women. Studies in animals have shown embryofoetal toxicity (see section 5.3). Human IgG4 is known to cross the placental barrier and nivolumab is an IgG4; therefore, nivolumab has the potential to be transmitted from the mother to the developing foetus. Nivolumab is not recommended during pregnancy and in women of childbearing potential not using effective contraception unless the clinical benefit outweighs the potential risk. Effective contraception should be used for at least 5 months following the last dose of nivolumab.
Breast-feeding It is unknown whether nivolumab is secreted in human milk. Because many medicinal products, including antibodies, can be secreted in human milk, a risk to the newborns/infants cannot be excluded. A decision must be made whether to discontinue breast-feeding or to discontinue from nivolumab therapy taking into account the benefit of breast-feeding for the child and the benefit of therapy for the woman.
Fertility Studies to evaluate the effect of nivolumab on fertility have not been performed. Thus, the effect of nivolumab on male and female fertility is unknown.
2.9 4.7 Effects on ability to drive and use machines
Nivolumab in combination with ipilimumab may have a minor influence on the ability to drive and use machines. Because of potential adverse reactions such as fatigue (see section 4.8), patients should be advised to use caution when driving or operating machinery until they are certain that nivolumab does not adversely affect them.
2.10 4.8 Undesirable effects
Please refer to the SmPCs of nivolumab and ipilimumab for the safety profile of nivolumab and ipilimumab used as monotherapy.
Nivolumab in combination with ipilimumab (see section 4.2) Summary of the safety profile When nivolumab is administered in combination with ipilimumab, refer to the Summary of Product Characteristics for ipilimumab prior to initiation of treatment.
MPM In the dataset of nivolumab 3 mg/kg in combination with ipilimumab 1 mg/kg in MPM (n = 300), with a minimum follow-up of 17.4 months, the most frequent adverse reactions (≥ 10%) were rash (25%), fatigue (22%), diarrhoea (21%), pruritus (16%), hypothyroidism (11%), and nausea (10%). The majority of adverse reactions were mild to moderate (Grade 1 or 2).
Tabulated summary of adverse reactions Adverse reactions reported in the pooled dataset for patients treated with nivolumab 3 mg/kg in combination with ipilimumab 1 mg/kg in MPM (n = 300) are presented in Table 2. These reactions are presented by system organ class and by frequency.
Frequencies are defined as: very common (≥ 1/10); common (≥ 1/100 to < 1/10); uncommon (≥ 1/1,000 to < 1/100); rare (≥ 1/10,000 to < 1/1,000); very rare (< 1/10,000), not known (cannot be estimated from available post-marketing data). Within each frequency grouping, adverse reactions are presented in the order of decreasing seriousness.
Table 2: Adverse reactions with nivolumab in combination with ipilimumab
| Immune system disorders | |
|---|---|
| Common | infusion-related reaction, hypersensitivity |
| Endocrine disorders | |
| Very common | hypothyroidism |
| Common | hyperthyroidism, adrenal insufficiency, hypophysitis, hypopituitarism |
| Uncommon | thyroiditis |
| Metabolism and nutrition disorders | |
| Common | decreased appetite |
| Nervous system disorders | |
| Uncommon | encephalitis |
| Cardiac disorders | |
| Uncommon | myocarditis |
| Respiratory, thoracic and mediastinal disorders | |
| Common | pneumonitis |
| Gastrointestinal disorders | |
| Very common | diarrhoea, nausea |
| Common | constipation, colitis, pancreatitis |
| Hepatobiliary disorders | |
| Common | hepatitis |
| Skin and subcutaneous tissue disorders | |
| Very common | rash (a), pruritus |
| Musculoskeletal and connective tissue disorders | |
| Common | musculoskeletal pain(b), arthritis |
| Uncommon | myositis |
| Renal and urinary disorders | |
| Common | acute kidney injury |
| Uncommon | renal failure |
| General disorders and administration site conditions | |
| Very common | fatigue |
| Investigations (c) | |
| Very common | increased AST, increased ALT, increased alkaline phosphatase, increased lipase, increased amylase, increased creatinine, hyperglycaemiad, lymphopaenia, anaemiae, hypercalcaemia, hypocalcaemia, hyperkalaemia, hypokalaemia, hyponatraemia, hypomagnesaemia |
| Common | increased total bilirubin, hypoglycaemia, leucopoenia, neutropaeniad,thrombocytopaenia, hypernatraemia, hypermagnesaemia |
(a) Rash is a composite term which includes maculopapular rash, rash erythematous, rash pruritic, rash follicular, rash macular, rash morbilliform, rash papular, rash pustular, rash papulosquamous, rash vesicular, rash generalised, exfoliative rash, dermatitis, dermatitis acneiform, dermatitis allergic, dermatitis atopic, dermatitis bullous, dermatitis exfoliative, dermatitis psoriasiform, drug eruption and pemphigoid.
(b) Musculoskeletal pain is a composite term which includes back pain, bone pain, musculoskeletal chest pain, musculoskeletal discomfort, myalgia, neck pain, pain in extremity, and spinal pain.
(c) Frequencies of laboratory terms reflect the proportion of patients who experienced a worsening from baseline in laboratory measurements. See “Description of selected adverse reactions; laboratory abnormalities” below.
(d) Life-threatening cases have been reported in completed or ongoing clinical studies.
(e) Anaemia is a composite term which includes, among other causes, haemolytic anaemia and autoimmune anaemia.
Description of selected adverse reactions
For further description of immune-related reactions, please refer to the SmPCs for nivolumab and ipilimumab.
Nivolumab in combination with ipilimumab is associated with immune-related adverse reactions. With appropriate medical therapy, immune-related adverse reactions resolved in most cases. Permanent discontinuation of treatment was required in a greater proportion of patients receiving nivolumab in combination with ipilimumab than in those receiving nivolumab monotherapy. Table 3 presents the percentage for immune-related adverse reactions who were permanently discontinued from treatment by dosing regimen. Additionally, for patients who experienced an event, Table 3 presents the percentage of patients who required high-dose corticosteroids (at least 40 mg daily prednisone equivalents) by dosing regimen. The management guidelines for these adverse reactions are described in section 4.4.
Table 3: Immune-related adverse reactions leading to permanent discontinuation or requiring high-dose corticosteroids by dosing regimen
| . | Nivolumab 3 mg/kg in combination with ipilimumab 1 mg/kg in MPM % |
|---|---|
| Immune-related adverse reaction leading to permanent discontinuation | . |
| Pneumonitis | 2.3 |
| Colitis | 5.0 |
| Hepatitis | 3.7 |
| Nephritis and renal Dysfunction | 1.3 |
| Endocrinopathies | 0.3 |
| Skin | 0.7 |
| Hypersensitivity/Infusion Reaction | 1.7 |
| Immune-related adverse reaction requiring high-dose corticosteroids (a),(b ) | |
| Pneumonitis | 70 |
| Colitis | 33 |
| Hepatitis | 42 |
| Nephritis and renal Dysfunction | 40 |
| Endocrinopathies | 10 |
| Skin | 8 |
| Hypersensitivity/Infusion Reaction | 17 |
(a) at least 40 mg daily prednisone equivalents (b) frequency is based on the number of patients who experienced the immune-related adverse reaction
Immune-related pneumonitis
In patients treated with nivolumab 3 mg/kg in combination with ipilimumab 1 mg/kg in MPM, the incidence of pneumonitis including interstitial lung disease was 6.7% (20/300). Grade 2 and Grade 3 cases were reported in 5.3% (16/300) and 0.7% (2/300) of patients, respectively. Median time to onset was 1.8 months (range: 0.3-20.8). Resolution occurred in 16 patients (80%) with a median time to resolution of 6.1 weeks (range: 1.1-113.1+).
Immune-related colitis In patients treated with nivolumab 3 mg/kg in combination with ipilimumab 1 mg/kg in MPM, the incidence of diarrhoea or colitis was 22.0% (66/300). Grade 2 and Grade 3 cases were reported in 7.3% (22/300) and 5.3% (16/300) of patients, respectively. Median time to onset was 3.9 months (range: 0.0-21.7). Resolution occurred in 66 patients (93.9%) with a median time to resolution of 3.1 weeks (range: 0.1-100.0+).
Immune-related hepatitis In patients treated with nivolumab 3 mg/kg in combination with ipilimumab 1 mg/kg in MPM, the incidence of liver function test abnormalities was 12.0% (36/300). Grade 2, Grade 3, and Grade 4 cases were reported in 1.7% (5/300), 4.3% (13/300), and 1.0% (3/300) of patients, respectively. Median time to onset was 1.8 months (range: 0.5-20.3). Resolution occurred in 31 patients (86.1%) with a median time to resolution of 4.1 weeks (range: 1.0-78.3+).
Immune-related nephritis and renal dysfunction In patients treated with nivolumab 3 mg/kg in combination with ipilimumab 1 mg/kg in MPM, the incidence of renal dysfunction was 5.0% (15/300). Grade 2 and Grade 3 cases were reported in 2.0% (6/300) and 1.3% (4/300) of patients, respectively. Median time to onset was 3.6 months (range: 0.5-14.4). Resolution occurred in 12 patients (80.0%) with a median time to resolution of 6.1 weeks (range: 0.9-126.4+).
Immune-related endocrinopathies In patients treated with nivolumab 3 mg/kg in combination with ipilimumab 1 mg/kg in MPM, the incidence of thyroid disorders was 14% (43/300). Grade 2 and Grade 3 thyroid disorders were reported in 9.3% (28/300) and 1.3% (4/300) of patients, respectively. Hypophysitis occurred in 2% (6/300) of patients. Grade 2 cases were reported in 1.3% (4/300) of patients. Grade 2 and Grade 3 hypopituitarism occurred in 1.0% (3/300) and 1.0% (3/300) of patients, respectively. Grade 2 and Grade 3 adrenal insufficiency occurred in 1.7% (5/300) and 0.3% (1/300) of patients, respectively. No cases of immune-related diabetes mellitus were reported. Median time to onset of these endocrinopathies was 2.8 months (range: 0.5-20.8). Resolution occurred in 17 patients (32.7%). Time to resolution ranged from 0.3 to 144.1+ weeks.
Immune-related skin adverse reactions In patients treated with nivolumab 3 mg/kg in combination with ipilimumab 1 mg/kg in MPM, the incidence of rash was 36.0% (108/300). Grade 2 and Grade 3 cases were reported in 10.3% (31/300) and 3.0% (9/300) of patients, respectively. Median time to onset was 1.6 months (range: 0.0-22.3). Resolution occurred in 71 patients (66.4%) with a median time to resolution of 12.1 weeks (range: 0.4-146.4+).
Rare cases of SJS and TEN some of them with fatal outcome have been observed (see sections 4.2 and 4.4).
Infusion reactions In patients treated with nivolumab 3 mg/kg in combination with ipilimumab 1 mg/kg in MPM, the incidence of hypersensitivity/infusion reactions was 12% (36/300); Grade 2 and Grade 3 cases were reported in 5.0% (15/300) and 1.3% (4/300) of patients, respectively.
Complications of allogeneic HSCT in classical Hodgkin Lymphoma Rapid onset of GVHD has been reported with nivolumab use before and after allogeneic HSCT (see section 4.4). In 49 evaluated patients from two cHL studies who underwent allogeneic HSCT after discontinuing nivolumab monotherapy, Grade 3 or 4 acute GVHD was reported in 13/49 patients (26.5%). Hyperacute GVHD, defined as acute GVHD occurring within 14 days after stem cell infusion, was reported in three patients (6%). A steroid-requiring febrile syndrome, without an identified infectious cause, was reported in six patients (12%) within the first 6 weeks post-transplantation, with three patients responding to steroids. Hepatic veno-occlusive disease occurred in one patient, who died of GVHD and multi-organ failure. Nine of 49 patients (18.4%) died from complications of allogeneic HSCT after nivolumab. The 49 patients had a median follow-up from subsequent allogeneic HSCT of 5.6 months (range: 0-19 months).
Laboratory abnormalities In patients treated with nivolumab 3 mg/kg in combination with ipilimumab 1 mg/kg in MPM, the proportion of patients who experienced a worsening from baseline to a Grade 3 or 4 laboratory abnormality was as follows: 2.4% for anaemia, 1.0% each for thrombocytopaenia and leucopoenia, 8.4% for lymphopaenia, 1.3% for neutropaenia, 3.1% for increased alkaline phosphatase, 7.1% each for increased AST and increased ALT, 1.7% for increased total bilirubin, 0.3% for increased creatinine, 2.8% for hyperglycaemia, 5.4% for increased amylase, 12.8% for increased lipase, 0.7% for hypernatraemia, 8.1% for hyponatraemia, 4.1% for hyperkalaemia, 2.0% for hypokalaemia, and 0.3% for hypocalcaemia.
Immunogenicity Of the patients who were treated with nivolumab in combination with ipilimumab and evaluable for the presence of anti-nivolumab antibodies, the incidence of anti-nivolumab antibodies was 26.0% with nivolumab 3 mg/kg and ipilimumab 1 mg/kg every 3 weeks, 25.7% with nivolumab 3 mg/kg every 2 weeks and ipilimumab 1 mg/kg every 6 weeks, and 37.8% with nivolumab 1 mg/kg and ipilimumab 3 mg/kg every 3 weeks. The incidence of neutralising antibodies against nivolumab was 0.5% with nivolumab 3 mg/kg and ipilimumab 1 mg/kg every 3 weeks, 0.7% with nivolumab 3 mg/kg every 2 weeks and ipilimumab 1 mg/kg every 6 weeks, and 4.6% with nivolumab 1 mg/kg and ipilimumab 3 mg/kg every 3 weeks. Of patients evaluable for the presence of anti-ipilimumab antibodies, the incidence of anti-ipilimumab antibodies ranged from 6.3 to 13.7% and neutralising antibodies against ipilimumab ranged from 0 to 0.4%.
Although the clearance of nivolumab was increased by 20% when anti-nivolumab-antibodies were present, there was no evidence of loss of efficacy or altered toxicity profile in the presence of nivolumab antibodies based on the pharmacokinetic and exposure-response analyses for combination.
Elderly No overall differences in safety were reported between elderly (≥ 65 years) and younger patients (< 65 years).
In MPM patients, there was a higher rate of serious adverse reactions and discontinuation rate due to adverse reactions in patients 75 years of age or older (68% and 35%, respectively) relative to all patients who received nivolumab in combination with ipilimumab (54% and 28%, respectively).
4.9 Overdose
No cases of overdose have been reported in clinical trials. In case of overdose, patients should be closely monitored for signs or symptoms of adverse reactions, and appropriate symptomatic treatment instituted immediately.
2.11 5. PHARMACOLOGICAL PROPERTIES
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: Antineoplastic agents, monoclonal antibodies. ATC code: L01XC17.
Mechanism of action Nivolumab is a human immunoglobulin G4 (IgG4) monoclonal antibody (HuMAb), which binds to the programmed death-1 (PD-1) receptor and blocks its interaction with PD-L1 and PD-L2. The PD-1 receptor is a negative regulator of T-cell activity that has been shown to be involved in the control of T-cell immune responses. Engagement of PD-1 with the ligands PD-L1 and PD-L2, which are expressed in antigen presenting cells and may be expressed by tumours or other cells in the tumour microenvironment, results in inhibition of T-cell proliferation and cytokine secretion. Nivolumab potentiates T-cell responses, including anti-tumour responses, through blockade of PD-1 binding to PD-L1 and PD-L2 ligands. In syngeneic mouse models, blocking PD-1 activity resulted in decreased tumour growth.
Combined nivolumab (anti-PD-1) and ipilimumab (anti-CTLA-4) mediated inhibition results in improved anti-tumour responses in metastatic melanoma. In murine syngeneic tumour models, dual blockade of PD-1 and CTLA-4 resulted in synergistic anti-tumour activity.
Clinical efficacy and safety
Malignant pleural mesothelioma
Randomised phase 3 study of nivolumab in combination with ipilimumab vs. chemotherapy (CA209743) The safety and efficacy of nivolumab 3 mg/kg every 2 weeks in combination with ipilimumab 1 mg/kg every 6 weeks were evaluated in a phase 3, randomised, open-label study (CA209743). The study included patients (18 years or older) with histologically confirmed and previously untreated malignant pleural mesothelioma of epithelioid or non-epithelioid histology, ECOG performance status 0 or 1, and no palliative radiotherapy within 14 days of first study therapy. Patients were enrolled regardless of their tumour PD-L1 status.
Patients with primitive peritoneal, pericardial, testis, or tunica vaginalis mesothelioma, interstitial lung disease, active autoimmune disease, medical conditions requiring systemic immunosuppression, and brain metastasis (unless surgically resected or treated with stereotaxic radiotherapy and no evolution within 3 months prior to inclusion in the study) were excluded from the trial. Randomisation was stratified by histology (epithelioid vs. sarcomatoid or mixed histology subtypes) and gender (male vs. female).
A total of 605 patients were randomised to receive either nivolumab in combination with ipilimumab (n = 303) or chemotherapy (n = 302). Patients in the nivolumab in combination with ipilimumab arm received nivolumab 3 mg/kg over 30 minutes by intravenous infusion every 2 weeks in combination with ipilimumab 1 mg/kg over 30 minutes by intravenous infusion every 6 weeks for up to 2 years. Patients in the chemotherapy arm received chemotherapy for up to 6 cycles (each cycle was 21 days). Chemotherapy consisted of cisplatin 75 mg/m2 and pemetrexed 500 mg/m2 or carboplatin 5 AUC and pemetrexed 500 mg/m2.
Treatment continued until disease progression, unacceptable toxicity, or for up to 24 months. Treatment could continue beyond disease progression if the patient was clinically stable and was considered to be deriving clinical benefit by the investigator. Patients who discontinued combination therapy because of an adverse reaction attributed to ipilimumab were permitted to continue nivolumab monotherapy. Tumour assessments were performed every 6 weeks after first dose of study treatment for the first 12 months, then every 12 weeks until disease progression or study treatment was discontinued.
CA209743 baseline characteristics were generally balanced across all treatment groups. The median age was 69 years (range: 25-89) with 72% ≥ 65 years of age and 26% ≥ 75 years of years. The majority of patients were white (85%) and male (77%). Baseline ECOG performance status was 0 (40%) or 1 (60%), 80% of patients with PD-L1 ≥ 1% and 20% with PD-L1 < 1%, 75% had epithelioid and 25% had non epithelioid histology.
The median duration of therapy in the nivolumab and ipilimumab arm was 5.55 months and 3.48 months in the chemotherapy arm. 23.7% of subjects received more than 12 months of nivolumab and ipilimumab treatment. 93.1% of subjects in the chemotherapy arm received all 6 cycles of chemotherapy.
CA209743 primary efficacy outcome measure was OS. PFS was initially a co-primary endpoint and later changed to a secondary endpoint through a protocol amendment. Additional efficacy endpoints were ORR, duration of response, and disease control rate (DCR) as assessed by Blinded Independent Central Review (BICR) utilising modified RECIST criteria.
The study demonstrated a statistically significant improvement in OS for patients randomised to nivolumab in combination with ipilimumab as compared to chemotherapy at the prespecified interim analysis when at least 403 events were observed (85% of the planned number of events for final analysis). Minimum follow-up for OS was 22 months. Efficacy results are shown in Figure 1 and Table 5.
Figure 1: Kaplan-Meier curves of OS (CA209743)
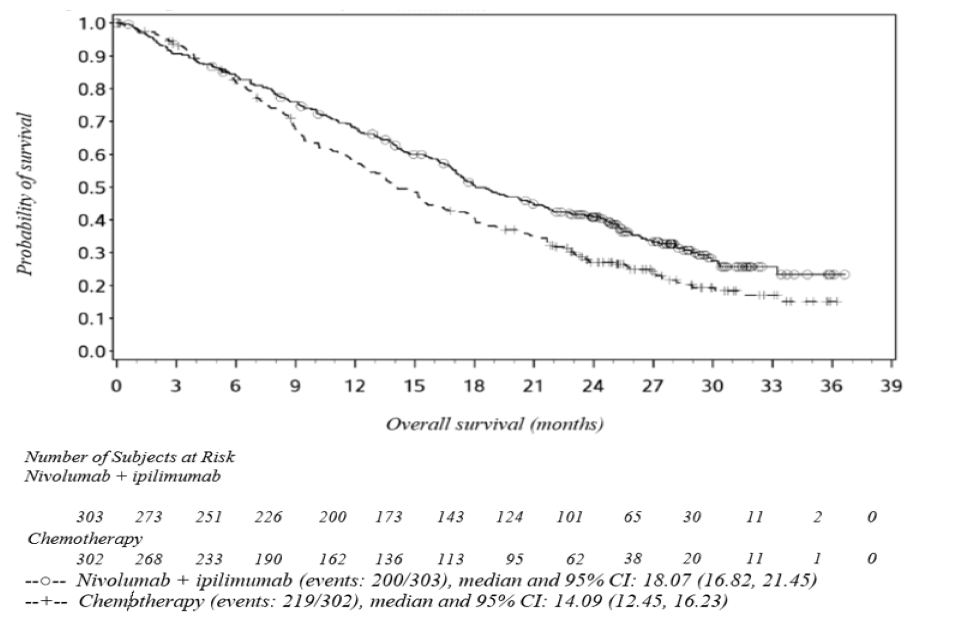
Table 5: Efficacy results (CA209743)

(a) Kaplan-Meier estimate. (b) Stratified Cox proportional hazard model. (c) p-value is compared with the allocated alpha of 0.0345 for this interim analysis.
Subsequent systemic therapy was received by 44.2% and 40.7% of patients in the combination and chemotherapy arms, respectively. Subsequent immunotherapy (including anti-PD-1, anti-PD-L1, and anti-CTLA-4) was received by 3.3% and 20.2% of patients in the combination and chemotherapy arms, respectively.
In study CA209743 prespecified subgroup analyses relative to chemotherapy, OS benefit was shown in patients treated with nivolumab in combination with ipilimumab with epithelioid histology (HR [95% CI] 0.85 [0.68, 1.06], n = 236) and in patients with non-epithelioid histology (HR [95% CI] 0.46 [0.31, 0.70], n = 67). OS benefit was also shown in patients with tumour PD-L1 expression < 1% (HR [95% CI] 0.94 [0.62, 1.40], n = 57) and tumour PD-L1 expression ≥ 1% (HR [95% CI] 0.69 [0.55, 0.87], n = 232).
Table 6: Overall Survival of Nivolumab + Ipilimumab vs Chemotherapy by Histology per IRT Source- All Randomised Subjects - CA209743
| Epithelioid (N = 471) | Non-epithelioid (N = 133) | |||
|---|---|---|---|---|
| Nivo+Ipi N = 236 | Chemo N = 235 | Nivo+Ipi N = 67 | Chemo N = 67 | |
| Overall Survival | ||||
| Median (95% CI), monthsa | 18.73 (17.05, 21.72) | 16.23 (14.09, 19.15) | 16.89 (11.83, 25.20) | 8.80 (7.62, 11.76) |
| HR (95% CI)(b) | 0.85 (0.68, 1.06) | 0.46 (0.31, 0.70) | ||
| Events, n | 157 | 164 | 43 | 55 |
(a) Based on Kaplan-Meier estimate (b) Unstratified Cox proportional hazards model
Abbreviations: chemo - chemotherapy, CI - confidence interval, HR - hazard ratio, ipi - ipilimumab, IRT - interactive response technology, n - number, nivo - nivolumab
PD-L1 expression was reported at a higher frequency in the non-epithelioid subgroup compared with the epithelioid subgroup in both the nivolumab + ipilimumab and chemotherapy arms. The magnitude of OS benefit comparing nivolumab + ipilimumab vs chemotherapy did vary according to histology and PD-L1 expression. Given the correlation between histology subtype and PD-L1 expression an efficacy subgroup analysis by histology and PD-L1 was performed. Results of the analysis are summarised in the table below.
Table 7: Subgroup Analysis by Histology and PD-L1 Expression Levels (≥ 1% and < 1%) - All PD-L1 Evaluable Subjects - CA209743
| Epithelioid | Non-epithelioid | |||
|---|---|---|---|---|
| PD-L1 < 1% Cut | PD-L1 ≥ 1% Cut | PD-L1 < 1% Cut | PD-L1 ≥ 1% Cut | |
| Subgroup Size (nivo+ipi vs chemo) | 47 vs 62 | 173 vs 162 | 10 vs 16 59 vs 57 | |
| Median OS, months (nivo+ipi vs chemo) | 17.3 vs 18.1 | 18.0 vs 15.5 | 15.7 vs 11.8 | 16.9 vs 7.7 |
| OS Rate, % (nivo+ipi vs chemo) | ||||
| 12 Month | 61.4 vs 70.3 | 71.2 vs 64.2 | 50.0 vs 40.0 | 65.5 vs 29.6 |
| 24 Month | 41.3 vs 29.2 | 41.6 vs 34.7 | 25.0 vs 6.7 | 38.5 vs 9.9 |
| OS HR (95% CI) | 0.99 (0.63, 1.56) | 0.81 (0.63, 1.06) | 0.69 (0.28, 1.67) | 0.43 (0.28, 0.66) |
| Median PFS, months (nivo+ipi vs chemo) | 4.2 vs 8.2 | 6.9 vs 7.7 | 3.5 vs 8.5 | 8.6 vs 5.3 |
| PFS HR (95%CI) | 1.91 (1.24, 2.94) | 0.99 (0.76, 1.28) | 1.12 (0.42, 2.95) | 0.46 (0.28, 0.74) |
| ORR, % (nivo+ipi vs chemo) | 21.3 vs 41.9 | 42.2 vs 50.0 | 20.0 vs 25.0 | 47.5 vs 28.1 |
| Median DOR, months (nivo+ipi vs chemo) | 8.3 vs 7.1 | 9.3 vs 6.7 | 10.5 vs 8.3 | 20.4 vs 4.2 |
Abbreviations: chemo - chemotherapy, CI - confidence interval, DOR - duration of response, HR - hazard ratio, ipi - ipilimumab, nivo - nivolumab, ORR - objective response rate, OS - overall survival, PD-L1 -programmed death ligand-1, PFS - progression-free survival
Safety and efficacy in elderly patients
No overall differences in safety or efficacy were reported between elderly (≥ 65 years) and younger patients (< 65 years).. Data from MPM patients showed a higher rate of serious adverse reactions and discontinuation rate due to adverse reactions in patients 75 years of age or older (68% and 35%, respectively) relative to all patients who received nivolumab in combination with ipilimumab (54% and 28%, respectively).
2.12 5.2 Pharmacokinetic properties
Nivolumab monotherapy
The pharmacokinetics (PK) of nivolumab is linear in the dose range of 0.1 to 10 mg/kg. The geometric mean clearance (CL), terminal half-life, and average exposure at steady state at 3 mg/kg every 2 weeks of nivolumab were 7.9 mL/h, 25.0 days, and 86.6 μg/mL, respectively, based on a population PK analysis.
The metabolic pathway of nivolumab has not been characterised. Nivolumab is expected to be degraded into small peptides and amino acids via catabolic pathways in the same manner as endogenous IgG.
Nivolumab in combination with ipilimumab
When nivolumab 1 mg/kg was administered in combination with ipilimumab 3 mg/kg, the CL of nivolumab was increased by 29% and the CL of ipilimumab was increased by 9%, which was not considered clinically relevant. When nivolumab 3 mg/kg was administered in combination with ipilimumab 1 mg/kg, the CL of nivolumab was increased by 1% and the CL of ipilimumab was decreased by 1.5%, which were not considered clinically relevant.
When administered in combination with ipilimumab, the CL of nivolumab increased by 20% in the presence of anti-nivolumab antibodies and the CL of ipilimumab increased by 5.7% in the presence of anti-ipilimumab antibodies. These changes were not considered clinically relevant.
Special populations
A population PK analysis suggested no difference in CL of nivolumab based on age, gender, race, solid tumour type, tumour size, and hepatic impairment. Although ECOG status, baseline glomerular filtration rate (GFR), albumin, body weight, and mild hepatic impairment had an effect on nivolumab CL, the effect was not clinically meaningful.
Renal impairment
The effect of renal impairment on the CL of nivolumab was evaluated in patients with mild (GFR < 90 and ≥ 60 mL/min/1.73 m2; n = 379), moderate (GFR < 60 and ≥ 30 mL/min/1.73 m2; n = 179), or severe (GFR < 30 and ≥ 15 mL/min/1.73 m2; n = 2) renal impairment compared to patients with normal renal function (GFR ≥ 90 mL/min/1.73 m2; n = 342) in population PK analyses. No clinically important differences in the CL of nivolumab were found between patients with mild or moderate renal impairment and patients with normal renal function. Data from patients with severe renal impairment are too limited to draw conclusions on this population (see section 4.2).
Hepatic impairment
The effect of hepatic impairment on the CL of nivolumab was evaluated in patients with mild hepatic impairment (total bilirubin 1.0 × to 1.5 × ULN or AST > ULN as defined using the National Cancer Institute criteria of hepatic dysfunction; n = 92) compared to patients with normal hepatic function (total bilirubin and AST ≤ ULN; n = 804) in the population PK analyses. No clinically important differences in the CL of nivolumab were found between patients with mild hepatic impairment and normal hepatic function. Nivolumab has not been studied in patients with moderate (total bilirubin > 1.5 × to 3 × ULN and any AST) or severe hepatic impairment (total bilirubin > 3 × ULN and any AST) (see section 4.2).
2.13 5.3 Preclinical safety data
Blockade of PD-L1 signalling has been shown in murine models of pregnancy to disrupt tolerance to the foetus and to increase foetal loss. The effects of nivolumab on prenatal and postnatal development were evaluated in monkeys that received nivolumab twice weekly from the onset of organogenesis in the first trimester through delivery, at exposure levels either 8 or 35 times higher than those observed at the clinical dose of 3 mg/kg of nivolumab (based on AUC). There was a dose-dependent increase in foetal losses and increased neonatal mortality beginning in the third trimester.
The remaining offspring of nivolumab-treated females survived to scheduled termination, with no treatment-related clinical signs, alterations to normal development, organ-weight effects, or gross and microscopic pathology changes. Results for growth indices, as well as teratogenic, neurobehavioral, immunological, and clinical pathology parameters throughout the 6-month postnatal period were comparable to the control group. However, based on its mechanism of action, foetal exposure to nivolumab may increase the risk of developing immune-related disorders or altering the normal immune response and immune-related disorders have been reported in PD-1 knockout mice.
Fertility studies have not been performed with Nivolumab.
3. 6. PHARMACEUTICAL PARTICULARS
3.1 6.1 List of excipients
Sodium citrate dihydrate
Sodium chloride
Mannitol (E421)
Pentetic acid (diethylenetriaminepentaacetic acid)
Polysorbate 80 (E433)
Sodium hydroxide (for pH adjustment)
Hydrochloric acid (for pH adjustment)
Water for injections
3.2 6.2 Incompatibilities
In the absence of compatibility studies, this medicinal product must not be mixed with other medicinal products. Nivolumab should not be infused concomitantly in the same intravenous line with other medicinal products.
3.3 6.3 Shelf life
Nivolumab
Unopened vial
3 years
After opening
From a microbiological point of view, once opened, the medicinal product should be infused or diluted and infused immediately.
After preparation of infusion
Chemical and physical in-use stability from the time of preparation has been demonstrated as follows (times are inclusive of the administration period):
| Infusion preparation | In-use stability | |
|---|---|---|
| Storage at 2ºC to 8ºC protected from light | Storage at room temperature (≤ 25°C) and room light | |
| Undiluted or diluted with sodium chloride 9 mg/mL (0.9%) solution for injection | 30 days | 24 hours (of total 30 days storage) |
| Diluted with 50 mg/mL (5%) glucose solution for injection | 24 hours | 8 hours (of total 24 hours storage) |
From a microbiological point of view the prepared solution for infusion, regardless of the diluent, should be used immediately. If not used immediately, in-use storage times and conditions prior to use are the responsibility of the user and would normally not be longer than 24 hours at 2°C to 8°C or 8 hours (of the total 24 hours of storage) at room temperature (≤ 25°C), unless infusion preparation has taken place in controlled and validated aseptic conditions.
Ipilimumab
Unopened vial
3 years
After opening
From a microbiological point of view, once opened, the medicinal product should be infused or diluted and infused immediately. The chemical and physical in-use stability of the undiluted or diluted concentrate (between 1 and 4 mg/ml) has been demonstrated for 24 hrs at 25°C and 2°C to 8°C. If not used immediately, the infusion solution (undiluted or diluted) may be stored for up to 24 hours in a refrigerator (2°C to 8°C) or at room temperature (20°C to 25°C).
3.4 6.4 Special precautions for storage
Store in a refrigerator (2°C-8°C).
Do not freeze.
Store in the original package in order to protect from light.
Nivolumab
The unopened vial can be stored at controlled room temperature up to 25°C with room light for up to 48 hours. For storage conditions after preparation of the infusion, see section 6.3.
Ipilimumab
For storage conditions after first opening or dilution of the medicinal product, see section 6.3.
3.5 6.5 Nature and contents of container
Nivolumab: 10 mL of concentrate in a 10 mL vial (Type I glass) with a stopper (coated butyl rubber) and a grey flip-off seal (aluminium). Pack size of 1 vial.
Ipilimumab: 40 ml of concentrate in a vial (Type I glass) with a stopper (coated butyl rubber) and a flip-off seal (aluminium). Pack size of 1.
3.6 6.6 Special precautions for disposal and other handling
Preparation should be performed by trained personnel in accordance with good practices rules, especially with respect to asepsis.
Preparation and administration of nivolumab
Calculating the dose
More than one vial of nivolumab concentrate may be needed to give the total dose for the patient.
Nivolumab in combination with ipilimumab in MPM
The prescribed dose of nivolumab for the patient is 360 mg given regardless of body weight (see section 4.2).
Preparing the infusion
Take care to ensure aseptic handling when you prepare the infusion.
Nivolumab can be used for intravenous administration either:
- without dilution, after transfer to an infusion container using an appropriate sterile syringe; or
- after diluting according to the following instructions:
- the final infusion concentration should range between 1 and 10 mg/mL.
- the total volume of infusion must not exceed 160 mL. For patients weighing less than 40 kg, the total volume of infusion must not exceed 4 mL per kilogram of patient weight.
Nivolumab concentrate may be diluted with either:
- sodium chloride 9 mg/mL (0.9%) solution for injection; or
- 50 mg/mL (5%) glucose solution for injection.
STEP 1
- Inspect the nivolumab concentrate for particulate matter or discoloration. Do not shake the vial. Nivolumab concentrate is a clear to opalescent, colourless to pale yellow liquid. Discard the vial if the solution is cloudy, is discoloured, or contains particulate matter other than a few translucent-to-white particles.
- Withdraw the required volume of nivolumab concentrate using an appropriate sterile syringe.
STEP 2
- Transfer the concentrate into a sterile, evacuated glass bottle or intravenous container (PVC or polyolefin).
- If applicable, dilute with the required volume of sodium chloride 9 mg/mL (0.9%) solution for injection or 50 mg/mL (5%) glucose solution for injection. For ease of preparation, the concentrate can also be transferred directly into a pre-filled bag containing the appropriate volume of sodium chloride 9 mg/mL (0.9%) solution for injection or 50 mg/mL (5%) glucose solution for injection.
- Gently mix the infusion by manual rotation. Do not shake.
Administration
Nivolumab infusion must not be administered as an intravenous push or bolus injection.
Administer the nivolumab infusion intravenously over a period of 30 minutes.
Nivolumab infusion should not be infused at the same time in the same intravenous line with other agents. Use a separate infusion line for the infusion.
Use an infusion set and an in-line, sterile, non-pyrogenic, low protein binding filter (pore size of 0.2 μm to 1.2 μm).
Nivolumab infusion is compatible with PVC and polyolefin containers, glass bottles, PVC infusion sets and in-line filters with polyethersulfone membranes with pore sizes of 0.2 µm to 1.2 µm.
After administration of the nivolumab dose, flush the line with sodium chloride 9 mg/mL (0.9%) solution for injection or 50 mg/mL (5%) glucose solution for injection.
Disposal
Do not store any unused portion of the infusion solution for reuse. Any unused medicinal product or waste material should be disposed of in accordance with local requirements.
Preparation and administration of ipilimumab
The recommended dose is 1mg/kg ipilimumab, in combination with 360mg nivolumab, for MPM (see section 4.2).
Please refer to the SmPC for ipilimumab for information on the preparation and administration of ipilimumab.
4. 7. SCIENTIFIC OPINION HOLDER
Bristol-Myers Squibb Pharmaceuticals Limited
Uxbridge Business Park Sanderson Road
Uxbridge UB8 1DH
United Kingdom
5. 8. EAMS NUMBER
15105/0013
6. 9. DATE OF SCIENTIFIC OPINION
DD/MM/YYYY
7. 10. DATE OF TEXT
DD/MM/YYYY
Additional information:
The prescribing oncologist should carefully read the information provided in the rest of this document. Each prescribing oncologist interested in enrolling a patient in the programme should contact Bristol-Myers Squibb (BMS) via email to EAMS@bms.com, including:
- Up-to-date Curriculum Vitae (stating GMC number and current institution),
- Postal address for receipt of documentation.
- Exact pharmacy details for shipment of drug (contact name, email address, phone and delivery address), and any names and email addresses of pharmacy team who will order drug (re-)supply;
- Name, email address and CV of other consultant oncologists who will be registering patients;
Patient details will need to be entered into an electronic database (Medidata RAVE), for which online training will be provided subsequent to registering interest in the scheme.
For NHS England only - additional requirement for registering a patient:
Following notification from BMS of eligibility approval, the HCP must complete a Blueteq form online and register their patient with NHS England, which is located at https://www.blueteq-secure.co.uk/Trust/default.aspx. Once the Blueteq form has been completed, an approval email will be received by the user and pharmacy stating the request has been approved, also stating an EAMS number.
An Informed Consent Form (ICF) will be provided by Bristol-Myers Squibb (BMS) via post, along with training materials, subsequent to registering interest in the scheme.
A Letter of Agreement (LoA) is required to be signed by the nominated oncologist and a legal representative from the trust. The LoA will be provided via email subsequent to registering interest in the scheme.
BMS will arrange training (including adverse event/safety information reporting training) and delivery of the programme materials, including the following:
Patient Alert Card
Copies of the Patient Alert Card will be provided with the programme materials. This will be given to all patients before starting treatment. It is a wallet sized Patient Alert Card to be carried at all times by the patient during treatment and for at least 5 months after completing treatment to show at all medical visits to HCPs other than the prescribers (e.g., emergency HCPs). It has contact details of the treating physician and it alerts other physicians that the patient is being treated with nivolumab and ipilimumab. It also contains important information on the main symptoms of the important adverse reactions and highlights the importance of notifying the treating physician immediately if symptoms occur, persist or worsen and also the importance of not attempting to self-treat any symptoms without consulting with an HCP first.
Drug Supply
BMS will check the eligibility of the patient and once the required documents which includes the Letter of Agreement that has been signed by the nominated oncologist and a legal representative from the Trust, an initial drug supply request can then be submitted by the prescriber in the Medidata RAVE system.
BMS will supply 6 weeks of EAMS drug supply for each drug request
For patients approved under this scheme and requiring ongoing drug supply, the prescribing oncologist (or designee registered in Medidata RAVE) will also be required to complete the drug re-supply eCRF page. The order should be placed 2 weeks before the next planned cycle is due.
The prescribing oncologist is requested to inform BMS if a patient discontinues treatment by completing the discontinuation details (within Medidata RAVE) with the patient’s last date of treatment.
Additional contact: Bristol-Myers Squibb Medical Information on 0800 731 1736 or medical.information@bms.com