Voxelotor: Treatment protocol: Information for healthcare professionals
Updated 25 October 2022

© Crown copyright 2022
This publication is licensed under the terms of the Open Government Licence v3.0 except where otherwise stated. To view this licence, visit nationalarchives.gov.uk/doc/open-government-licence/version/3 or write to the Information Policy Team, The National Archives, Kew, London TW9 4DU, or email: psi@nationalarchives.gov.uk.
Where we have identified any third party copyright information you will need to obtain permission from the copyright holders concerned.
This publication is available at https://www.gov.uk/government/publications/voxelotor-in-the-treatment-of-sickle-cell-disease/voxelotor-treatment-protocol-information-for-healthcare-professionals
This protocol has been revised to include a new email address for adverse drug reaction reporting to the scientific opinion holder.
Introduction
The aim of the Early Access to Medicines Scheme (EAMS) is to provide earlier availability of promising new unlicensed medicines and medicines used outside their licence, to UK patients that have a high unmet clinical need. The medicinal products included in the scheme are those that are intended to treat, diagnose or prevent seriously debilitating or life-threatening conditions where there are no adequate treatment options. More information about the scheme can be found here: http://www.mhra.gov.uk/Howweregulate/Innovation/EarlyaccesstomedicinesschemeEAMS/index.htm
This information is intended for healthcare professionals and is provided by the pharmaceutical company that manufactures the EAMS medicine. This medicine does not yet have a licence (marketing authorisation) and the information is provided to assist physicians in prescribing this unlicensed medicine. Guidance on prescribing unlicensed medicines can be found on the GMC webpage: https://www.gmc-uk.org/guidance/ethical_guidance/14327.asp
The scientific opinion is based on assessment of the information supplied to the MHRA on the benefits and risks of this promising new medicine. As such, this is a scientific opinion and should not be regarded as a medicine licensed by the MHRA or a future commitment by the MHRA to license such a medicine, nor should it be regarded as an authorisation to sell or supply such a medicine. A positive scientific opinion is not a recommendation for use of the medicine and should not be interpreted as such. Under EAMS the risk and legal responsibility for prescribing a ‘special’ remains with the physician, and the opinion and EAMs documentation published by the MHRA are intended only to inform physicians’ decision making and not to recommend use. An EAMS scientific opinion does not affect the civil liability of the manufacturer or any physician in relation to the product.
Healthcare professionals should also refer to the summary information on the pharmacovigilance system which is provided in the document ‘Early Access to Medicines Scheme – Treatment protocol – Information on the pharmacovigilance system’.
Scientific opinion period: The MHRA will withdraw the EAMS positive scientific opinion when a marketing authorisation (drug licence) is issued for the EAMS product covering the EAMS indication, or if following scientific assessment, the EAMS criteria are considered to be no longer met.
Treatment protocol update(s): In case of substantial new efficacy or safety data, the treatment protocol may need to be updated.
Contact information regarding queries on using this EAMS medicine can be found at the end of this document.
Information for healthcare professionals
1. NAME OF THE MEDICINAL PRODUCT
Voxelotor
2. QUALITATIVE AND QUANTITATIVE COMPOSITION
Each film-coated tablet contains 500 mg of voxelotor.
For the full list of excipients, see section 6.1.
3. PHARMACEUTICAL FORM
Film-coated tablet.
Light yellow to yellow, oval shaped, biconvex, film-coated tablet of approximately 18 mm × 10 mm, debossed with “GBT 500” on one side.
4. CLINICAL PARTICULARS
4.1 EAMS therapeutic indication
Voxelotor is indicated for the treatment of haemolytic anaemia (haemoglobin ≤ 10.5 g/dL) due to sickle cell disease (SCD) in adults and paediatric patients 12 years of age and older as monotherapy or in combination with hydroxycarbamide.
Patients ineligible for participation in this early access to medicines scheme (EAMS) include : those with severe hepatic impairment, those with severe renal impairment or end-stage renal disease (ESRD) requiring dialysis, female patients who are pregnant or breastfeeding, patients who have medical, psychological, or behavioural conditions, which, in the opinion of the treating physician, makes patient unsuitable to participate in this EAMS or patients who have participated in a clinical trial of an investigational agent (or medical device) within 30 days of participation in EAMS.”
4.2 Posology and method of administration
Treatment should be initiated by physicians experienced in the management of SCD.
Posology
The recommended dose of voxelotor is 1,500 mg (three 500 mg film-coated tablets) taken orally once daily.
If a dose is missed, treatment should be continued on the day following the missed dose.
Paediatric population
The recommended dose of voxelotor in patients 12 to < 18 years of age is the same as for adults.
The safety and efficacy of voxelotor in paediatric patients below the age of 12 years have not been established yet. No data are available.
Special populations
Renal impairment
No dose adjustment is recommended in patients with mild or moderate renal impairment. Patients with severe renal impairment or ESRD requiring dialysis are excluded from this EAMS (see section 4.4).
Hepatic impairment
No dose adjustment of voxelotor is recommended for patients with mild or moderate hepatic impairment. Patients with severe hepatic impairment are excluded from this EAMS (see section 4.4).
Method of administration
Voxelotor film-coated tablets should be swallowed whole with water. voxelotor can be taken with or without food (see section 5.2). Tablets should not be cut, crushed, or chewed because of the unpleasant taste.
4.3 Contraindications
Hypersensitivity to the active substance or to any of the excipients listed in section 6.1. (see section 4.4.).
4.4 Special warnings and precautions for use
Hypersensitivity reactions
Serious hypersensitivity reactions have been observed in < 1% of patients treated with voxelotor in clinical studies. Clinical manifestations may include generalized rash, urticaria, mild shortness of breath, mild facial swelling, and eosinophilia (see section 4.8).
If hypersensitivity reactions occur, voxelotor must be discontinued and appropriate medical therapy must be administered. Voxelotor must not be reinitiated in patients who experience these symptoms with previous use.
Laboratory test interference
Voxelotor administration may interfere with measurement of haemoglobin (Hb) subtypes (HbA, HbS, and HbF) by high-performance liquid chromatography (HPLC). If precise quantitation of Hb species is required, chromatography should be performed when the patient has not received voxelotor therapy in the immediately preceding 10 days.
Renal impairment
No clinically significant differences in the pharmacokinetics of voxelotor were observed in subjects without SCD with mild to severe renal impairment (see section 5.2). Safety of voxelotor has not been evaluated in patients with ESRD requiring dialysis. SCD patients with severe renal impairment or ESRD requiring dialysis are excluded from this EAMS.
Hepatic impairment
There are limited data on safety of voxelotor in patients with SCD with different degrees of hepatic impairment. Based on pharmacokinetic data in subjects without SCD, severe hepatic impairment increases voxelotor exposures (see section 5.2). Patients with severe hepatic impairment are excluded from this EAMS.
Concomitant strong CYP3A4 inducers
Concomitant use of strong CYP3A4 inducers with voxelotor should be avoided due to the risk of decreased efficacy of voxelotor (see section 4.5).
SCD genotypes
Most patients (90.5%) in the pivotal Phase 3 study had SCD genotype HbSS (75.2%) or HbS/β0 thalassemia (15.3%). Therefore, safety and efficacy data on other SCD genotypes are limited.
Elderly
Clinical studies of voxelotor did not include patients > 65 years of age. In the absence of data, voxelotor should be used with caution in this population after careful consideration of the potential benefit/risk on an individual basis.
Combination therapy with hydroxycarbamide
When voxelotor is administered in combination with hydroxycarbamide, the prescribing information of hydroxycarbamide should be consulted.
Immunosuppressive effects
Voxelotor decreased the humoral immune response to antigens in both rats and monkeys. These changes were not considered adverse. Clinical relevance in already immunocompromised patients or in patients treated with immunosuppressive drugs cannot be excluded.
Excipients
This medicinal product contains less than 1 mmol sodium (23 mg) per 1,500 mg (daily dose), that is to say essentially ‘sodium-free’.
4.5 Interaction with other medicinal products and other forms of interaction
Effect of other medicinal products on voxelotor
Strong CYP3A4 inducers
Coadministration of strong CYP3A4 inducers may decrease voxelotor exposures and may lead to reduced efficacy.
Coadministration of voxelotor with strong CYP3A4 inducers (i.e., rifampicin, phenobarbital, carbamazepine, phenytoin, and St John’s wort extract) should be avoided.
Other interactions studied
Itraconazole (a strong CYP3A4 inhibitor), omeprazole (acid reducing agent), and hydroxycarbamide had no effect on the pharmacokinetics of voxelotor.
Effect of voxelotor on other medicinal products
CYP3A4 Substrates
Voxelotor increased the systemic exposure of midazolam (a sensitive CYP3A4 substrate). The observed exposure increase of the CYP3A4 substrate midazolam was 1.6-fold in healthy volunteers at a voxelotor sub therapeutic dose (observed voxelotor Cmax 7.0 - 8.0 microgram/mL and AUC 126.3 - 148.9 microgram·hr/mL). The effect at the full dose level of voxelotor is expected to be larger. Coadministration of voxelotor with sensitive CYP3A4 substrates with a narrow therapeutic index (i.e., alfentanil, sirolimus, and tacrolimus) should be avoided. If concomitant use is unavoidable, consider dose reduction of the sensitive CYP3A4 substrate(s).
CYP2B6 substrates
In vitro studies indicated that voxelotor acts as an inhibitor and inducer of CYP2B6 (see section 5.2). The clinical relevance is currently unknown and, caution is recommended when co-administering voxelotor with sensitive substrates of CYP2B6 such as bupropion and efavirenz.
CYP2C8, CYP2C9, and CYP2C19 Substrates
Voxelotor is an in vitro inhibitor of CYP2C8, CYP2C9, and CYP2C19 at maximal systemic concentrations. There was no observed change on the exposures of the S-warfarin (CYP2C9 substrate) and omeprazole (CYP2C19 substrate) in healthy volunteers at a sub therapeutic voxelotor dose (observed voxelotor Cmax 7.0 - 8.0 microgram/mL and AUC 126.3 - 148.9 microgram·hr/mL). The effect at the full dose level of voxelotor is currently unknown. Caution is recommended when co administering voxelotor with sensitive substrates of CYP enzymes.
Transporter-mediated drug interactions
In vitro studies indicated that voxelotor may act as an inhibitor of OATP1B1, OAT3 and MATE1 transporters (see section 5.2). Therefore, caution is recommended when co-administering voxelotor with sensitive substrates of these transporters, especially for those substrates with a narrow therapeutic index.
Concomitant use of voxelotor with digoxin (a P-gp substrate) did not alter digoxin to a clinically relevant extent. Voxelotor is not an inhibitor of bile salt export pump (BSEP). It is not known if voxelotor affects the oral absorption of BCRP substrates.
Oral contraceptives and other steroidal agents
Specific interaction studies with oral contraceptives have not been performed. However, based on the results of in vitro studies, a negative impact of voxelotor on contraceptive efficacy is not expected.
Other interactions studied
Voxelotor did not change the systemic exposure of caffeine (CYP1A2 substrate) and metoprolol (CYP2D6 substrate).
4.6 Fertility, pregnancy and lactation
Pregnancy
There are no or limited amount of data from the use of voxelotor in pregnant women. Animal studies do not indicate direct or indirect harmful effects with respect to reproductive toxicity (see section 5.3). As a precautionary measure, it is preferable to avoid the use of voxelotor during pregnancy.
Breast-feeding
It is unknown whether voxelotor/metabolites are excreted in human milk. Available pharmacokinetic/toxicological data in animals have shown excretion of voxelotor in milk and subsequent uptake in pups (for details see section 5.3). A risk to the newborns/infants cannot be excluded. Voxelotor should not be used during breast-feeding.
Fertility
No human data are available on the effect of voxelotor on fertility. In rats, effects on sperm motility and morphology were observed. These effects did not, however, affect the reproductive performance (see section 5.3). Relevance to human is not known.
4.7 Effects on ability to drive and use machines
Voxelotor has no or negligible influence on the ability to drive and use machines.
4.8 Undesirable effects
Summary of the safety profile
The most common adverse reactions include headache (31.8%), diarrhoea (22.7%) and abdominal pain (22.7%). Serious adverse reactions include headache (1.1%) and drug hypersensitivity (1.1%). Permanent discontinuation due to an adverse reaction occurred in 2.3% of patients.
Dose modifications (dose reduction or dosing interruption) due to an adverse reaction occurred in 13.6% of patients who received voxelotor in the pivotal study. The adverse reactions requiring dose modification included rash (4.5%), diarrhoea (3.4%), headache (2.3%), nausea (2.3%), abdominal pain (1.1%), and drug hypersensitivity (1.1%).
Tabulated list of adverse reactions
Table 1 lists adverse drug reactions that occurred in patients treated with voxelotor 1,500 mg during a 72-week, randomized, double-blind, placebo-controlled pivotal Phase 3 study (n=88).
Adverse reactions reported with voxelotor are listed by system organ class and preferred term. Within each system organ class, adverse reactions are listed under frequency categories. Frequencies are defined as very common (≥ 1/10); common (≥ 1/100 to < 1/10); uncommon (≥ 1/1,000 to < 1/100); rare (≥ 1/10,000 to < 1/1,000); very rare (< 1/10,000); not known (cannot be estimated from available clinical study data). Within each frequency grouping, adverse reactions are presented in the order of decreasing seriousness.
Table 1: Adverse reactions
| System organ class | Adverse reactions[footnote 1] | Frequency category |
| Immune system disorders | Drug hypersensitivity | Uncommon |
| Nervous system disorders | Headache | Very common |
| Gastrointestinal disorders | Diarrhoea, Abdominal pain[footnote 2], Nausea | Very common |
| Skin and subcutaneous tissue disorders | Rash[footnote 3] | Very common |
Description of selected Adverse Reactions
Gastrointestinal (GI) disorder
In the pivotal Phase 3 study, the most commonly reported GI adverse reactions were diarrhoea, abdominal pain and nausea with diarrhoea and nausea showing a dose-dependent effect. The majority of reported GI events were Grade 1 or 2 and were manageable without the need for dose interruption, reduction or treatment discontinuation and resolved with continued use. Gastrointestinal adverse reactions resulting in dose reductions occurred in 4.5% of patients. Diarrhoea was the most common adverse reaction and was reported in 22.7%, and 11.0% of patients in the voxelotor 1,500 mg, and placebo groups respectively. There was 1 (1.1%) report of Grade 3 diarrhoea. A serious adverse reaction of nausea resulting in hospitalisation occurred in 1 (1.1%) patient in the voxelotor 1,500 mg group.
Drug hypersensitivity
In the pivotal Phase 3 study, 1 patient (1.1%) experienced drug hypersensitivity on Study Day 40. Observed symptoms included generalized morbilliform rash, urticaria, mild shortness of breath, mild facial swelling, pyrexia, headache, and diarrhoea. Elevated eosinophils were noted. Symptoms abated after voxelotor was withheld, and recurrence was observed after reintroduction of voxelotor. Event resolved with antihistamine and oral corticosteroids.
Rash
In the pivotal Phase 3 Study, rash was reported in 14.8% and 11.0% of patients in the voxelotor 1,500 mg and placebo groups, respectively. The majority of rash events were similar in appearance (consistent with typical maculopapular drug eruptions) and distribution, were not associated with extradermal symptoms, and were clinically manageable with or without treatment including oral antihistamines or topical corticosteroids. Exposure-response analysis did not reveal a statistically significant dose- or exposure-response relationship.
Paediatric population
The safety profile observed in paediatric patients 12 to < 18 years of age treated with voxelotor in the clinical studies was similar to that seen in adult patients.
Reporting of suspected adverse reactions
Reporting suspected adverse reactions to the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the Yellow Card Scheme at: www.mhra.gov.uk/yellowcard or search for MHRA Yellow Card in the Google Play or Apple App Store.
As a minimum, all serious adverse events (SAEs) regardless of causality should be reported to the manufacturer of the product, within 24 hours of event knowledge and no later than one business day.
Additionally, any other adverse events and reports of Pregnancy should be reported to: globalbloodtherapeutics@parexel.com
4.9 Overdose
There was one report of overdose in the pivotal Phase 3 study where a patient took a total of 3,000 mg of voxelotor at one time. There were no adverse reactions associated with this event.
In the event of an overdose, the patient should be treated symptomatically, and supportive measures instituted as required.
5. PHARMACOLOGICAL PROPERTIES
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: Other haematological agents, ATC code: B06AX03
Mechanism of action
Voxelotor is a haemoglobin S (HbS) polymerisation inhibitor that binds to HbS with a 1:1 stoichiometry and exhibits preferential partitioning to red blood cells (RBCs). By increasing the affinity of Hb for oxygen, voxelotor demonstrates dose-dependent inhibition of HbS polymerisation. Voxelotor inhibits RBC sickling and improves RBC deformability.
Pharmacodynamic effects
The pharmacodynamic effect of voxelotor treatment demonstrated a dose-dependent increase in Hb oxygen affinity as determined by the change in p20 and p50 (partial pressure of oxygen at which Hb oxygen saturation of 20% or 50% is achieved) that was linearly correlated with voxelotor exposure leading to inhibition of HbS polymerisation. The impact of the anti-polymerisation effect is to reduce measures of haemolysis (indirect bilirubin) with a concomitant decrease in percent reticulocyte count and an increase in Hb consistent with improvement in haemolytic anaemia.
Cardiac electrophysiology
At plasma concentrations approximately 2-fold above therapeutic concentrations, voxelotor does not prolong QT interval to any clinically relevant extent.
Clinical efficacy and safety
The efficacy and safety of voxelotor in patients with SCD was evaluated in a randomised, double-blind, placebo-controlled, multicentre study (EudraCT2016-003370-40). In this study, 274 patients were randomised to daily oral administration of voxelotor 1,500 mg (N=90), voxelotor 900 mg (N=92), or placebo (N=92). Patients were included if they had baseline Hb ≥ 5.5 g/dL (3.41 mmol/L) to ≤ 10.5 g/dL (6.52 mmol/L) and 1 to 10 vaso-occlusive crisis (VOC) events within 12 months prior to enrolment. Otherwise eligible patients on stable doses of hydroxycarbamide for at least 90 days were allowed to continue hydroxycarbamide therapy throughout the study. Randomization was stratified by patients already receiving hydroxycarbamide (yes, no), geographic region (North America, Europe, Other), and age (12 to < 18 years, 18 to 65 years). Key exclusion criteria included patients who (1) were receiving regular RBC transfusions, (2) received RBC transfusions within 60 days, (3) received erythropoietin within 28 days of enrolment, (4) had known active hepatitis A, B, or C or who were known to be human immunodeficiency virus (HIV) positive (5) had severe renal insufficiency, (6) had uncontrolled liver disease, (7) were pregnant, or (8) were breast-feeding.
Seventy-five percent of patients had HbSS genotype, 15% had HbS/β0-thalassemia, 4% HbS/β+ thalassemia, 3% HbSC, and 3% other sickle cell variants. The majority were receiving hydroxycarbamide therapy (65%). The median age was 24 years (range: 12 to 64 years); 46 (17%) patients were 12 to < 18 years of age. Median baseline Hb was 8.5 g/dL (5.28 mmol/L) (5.9 to 10.8 g/dL [3.66 to 6.70 mmol/L]). One hundred fifteen (42%) had 1 VOC event and 159 (58%) had 2 to 10 events within 12 months prior to enrolment. Of the 274 patients, 75 (27.4%) discontinued the study early. The main reasons for discontinuation were withdrawal of consent (10.2%) and adverse events (8.4%).
Efficacy was based on the following primary endpoint: Hb response rate defined as a Hb increase of > 1 g/dL (0.62 mmol/L) from baseline to Week 24 in patients treated with voxelotor 1,500 mg versus placebo. The response rate for voxelotor 1,500 mg was 51.1% (46/90) compared to 6.5% (6/92) in the placebo group (p < 0.001). No outlier subgroups were observed (Figure 1). The increase in Hb was observed beginning at Week 2 and maintained through Week 72. The distribution of Hb change from baseline for individual patients completing 24 weeks of treatment with voxelotor 1,500 mg or placebo is depicted in Figure 2.
Figure 1: Haemoglobin response at Week 24 by subgroup (voxelotor 1,500 mg vs placebo) (intent-to-treat [ITT] population)
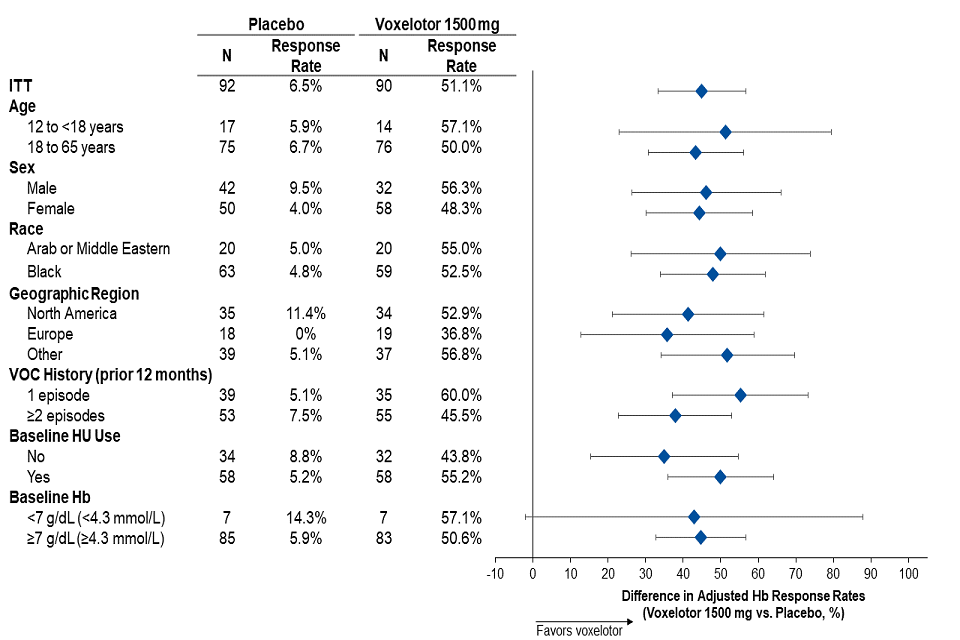
Figure 2: Subject-level change from baseline in haemoglobin at Week 24 in patients who completed 24 weeks of treatmenta,b
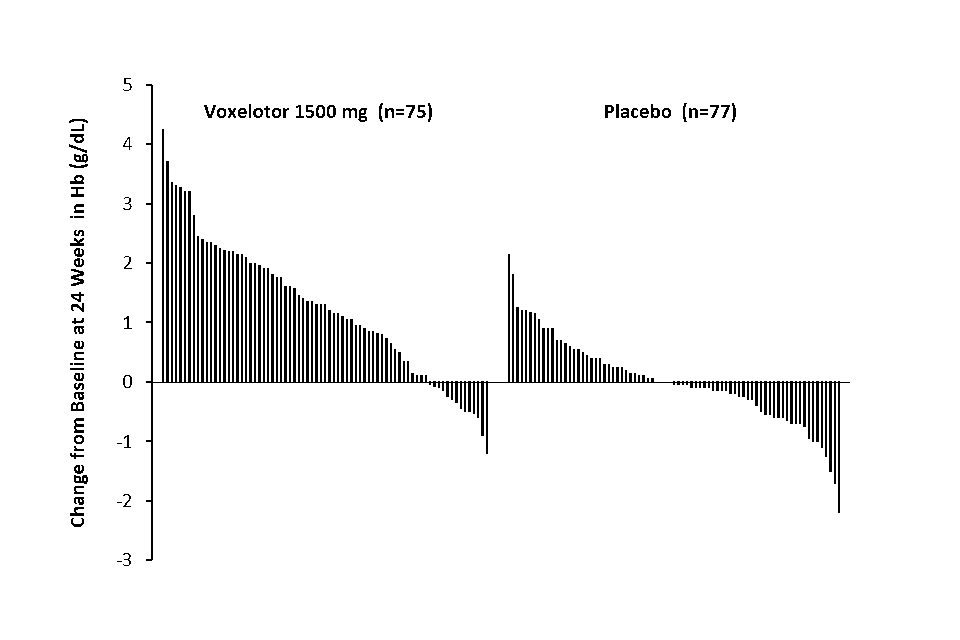
a. Approximately 83% of all randomised patients completed 24 weeks of treatment.
b. In the International System of Units (SI), the Hb range of -3 to 5 g/dL on the Y axis equates to -1.86 mmol/L to 3.10 mmol/L based on a conversion factor of 0.6206.
Additional efficacy evaluation included change in Hb and percent change in indirect bilirubin and percent reticulocyte count from baseline to Week 24 and Week 72 (Table 2).
Table 2: Adjusted mean (SE) change from baseline to Weeks 24 and 72 in haemoglobin and clinical measures of haemolysis (ITT population)
| Week 24 Voxelotor 1,500 mg QD (N=90) | Week 24 Placebo (N=92) | Week 72 Voxelotor 1,500 mg QD (N=90) | Week 72 Placebo (N=92) | |
| Haemoglobin g/dL | 1.13 (0.13) | -0.10 (0.13) | 1.02 (0.15) | 0.02 (0.15) |
| Haemoglobin mmol/L | 0.70 (0.08) | -0.06 (0.08) | 0.63 (0.09) | 0.01 (0.09) |
| P-value | < 0.001 | < 0.001 | < 0.001 | < 0.001 |
| Indirect Bilirubin % | -29.1 (3.5) | -2.8 (3.5) | -23.9 (4.9) | 2.7 (4.9) |
| Percent Reticulocyte Count % | -18.0 (4.7) | 6.8 (4.7) | -7.6 (5.5) | 11.0 (5.5) |
SE = standard error
The total number and annualized incidence rate (IR) of on-treatment VOCs were as follows: 219 events with adjusted IR of 2.4 events/year in the voxelotor 1,500 mg group and 293 events with adjusted IR of 2.8 events/year in the placebo group. No statistically significant difference was observed between the treatment groups; however, the study was not designed to detect a difference.
Treatment with voxelotor 1,500 mg resulted in improved clinician-reported patient outcomes as assessed using the Clinical Global Impression of Change (CGI-C).
Based on the CGI-C scale, 59.3% (32/54) of the patients in the voxelotor 1,500 mg group and 43.6% (24/55) of the patients in the placebo group were assessed by study medical personnel familiar with the patient’s baseline status as having moderately or very much improved at Week 24 relative to baseline. At Week 72, 73.6% (39/53) of the patients in the voxelotor 1,500 mg group and 47.1% (24/51) of the patients in the placebo group were assessed as having moderately or very much improved relative to baseline (p < 0.01; Figure 3).
Figure 3: Clinical global impression of change, moderately or very much improved by visit (ITT population)
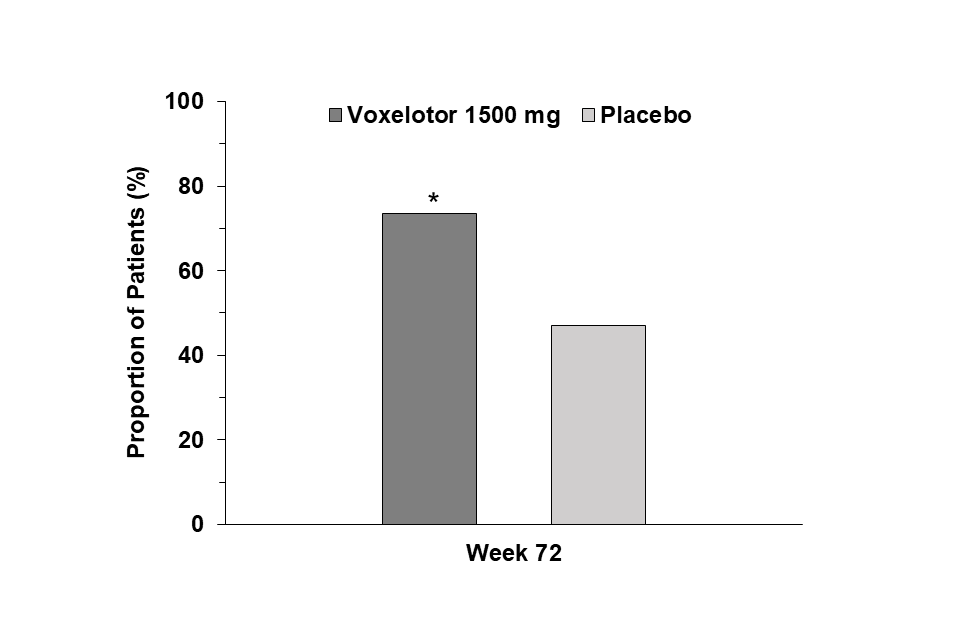
*p < 0.01 (voxelotor 1,500 mg vs placebo).
In the pivotal study leg ulcers were observed at baseline: 4 in the voxelotor 1,500 mg group, 3 in the placebo group. In the voxelotor group, all 4 patients with leg ulcers at baseline, improved after treatment (3 patients had resolution by Week 72 and 1 patient with moderate severity at baseline improved to mild). One patient developed new leg ulcers during treatment. In contrast, in the placebo group, only 1 of the 3 patients with leg ulcers at baseline had improvement and 5 patients developed new leg ulcers.
Paediatric population
Study GBT440 007
Study GBT440 007 is an ongoing Phase 2, multicentre, open-label single- and multiple-dose study designed to evaluate the safety, tolerability, PK, and efficacy of voxelotor in paediatric patients with SCD. Efficacy and safety data from the completed multiple-dose part in patients 12 to < 18 years of age with SCD (HbSS or HbS/β0-thalassemia) who received voxelotor 900 mg or 1,500 mg for 24 weeks are discussed here.
In total, 25 patients received voxelotor 900 mg and 15 patients received voxelotor 1,500 mg. The median age in the voxelotor 1,500 mg group was 14 years (range: 12-17 years), 33% were male and 73% were Black. Most patients in the 1,500 mg group had HbSS genotype (80%) and all used hydroxycarbamide at baseline. Thirty-three percent (33%) had no history of VOC within the 12 months prior to screening and 33% had 1 or 2 VOCs in the 12 months prior to screening. Median baseline Hb level was 8.8 g/dL (5.46 mmol/L). Eighty-eight percent (88.0%) of patients in the voxelotor 900 mg group and 80.0% of patients in the voxelotor 1,500 mg group completed the study with 24 weeks of dosing. One patient in the voxelotor 1,500 mg group discontinued due to an adverse reaction (Grade 1 diarrhoea).
Efficacy assessments included clinical measures of anaemia (Hb) and haemolysis (percent reticulocyte count and indirect bilirubin). Consistent with the results of the Phase 3 Study, voxelotor, improvements in Hb were observed as early as Week 2 and were maintained through Week 24: median change in Hb from baseline to Week 20/Week 24 average was 0.7 g/dL (0.43 mmol/L) for the 1,500 mg group, decrease in percent reticulocyte count at 24 weeks was -17.4% (-35.6, -36.5) and decrease in indirect bilirubin was -42.8% ( 50.5, -15.4) in the voxelotor 1,500 mg group. The safety profile was consistent with that observed in the Phase 3 Study.
5.2 Pharmacokinetic properties
Absorption
The median plasma and whole blood Tmax of voxelotor after oral administration is 2 hours. The mean peak concentrations in whole blood and RBCs are observed between 6 and 18 hours after oral administration. The PK are linear over the dose range of 100 mg to 2800 mg. Steady-state after repeated administration is reached within 8 days and exposures of voxelotor plasma and whole blood (Table 3) are consistent with accumulation predicted based on single dose data in patients with SCD.
Table 3: Pharmacokinetics parameters of voxelotor in plasma and whole blood (Subjects with SCD)
| PK parameter | Voxelotor 1,500 mg Geometric mean (%CV) |
| Plasma PK | |
| AUC0-24h (microgram·hr/mL) | 278 (28.4) |
| Cmax (microgram/mL) | 14 (24.5) |
| Half-life (hours) | 38.7 (30.2) |
| Whole Blood PK | |
| AUC0-24h (microgram·hr/mL) | 3830 (33.5) |
| Cmax (microgram/mL) | 180 (31) |
Effect of food
In healthy subjects, administration of a single 900 mg dose of voxelotor with a high-fat meal resulted in a 45% and 42% increase in whole blood Cmax and AUC, respectively, compared to fasted conditions.
In clinical studies, subjects with SCD took voxelotor without instructions with respect to food intake and had plasma and whole blood voxelotor exposures similar to subjects with SCD who took voxelotor after an overnight fast. The difference is less than 20% for any of the parameters and not considered to be clinically significant. Therefore, voxelotor can be taken with or without food.
Distribution
Voxelotor is absorbed into plasma and is then distributed predominantly into RBCs due to its preferential binding to Hb. Voxelotor apparent volume of distribution of the central compartment and peripheral compartment in patients with SCD are 333 L and 72.3 L in plasma, respectively. Protein binding is 99.8% in vitro. The blood-to-plasma ratio is approximately 15:1 in patients with SCD.
The pharmacokinetics of voxelotor in healthy subjects is different from patients with SCD due to the differences in blood-to-plasma partitioning (ratio 32:1). The volume of distribution in healthy subjects is approximately 754 L.
Biotransformation
In vitro and in vivo studies indicate that voxelotor is extensively metabolized through Phase I (oxidation and reduction), Phase II (glucuronidation) and combinations of Phase I and II metabolism. Oxidation of voxelotor is mediated primarily by CYP3A4, with minor contribution from CYP2C19, CYP2B6, and CYP2C9. Sulfatation of voxelotor is mediated primarily by SULT1B1 and SULT1C4 and direct glucuronidation of voxelotor is mediated by UGT1A1 and UGT1A9. The major plasma metabolite results from O-dealkylation-sulfation and represents 16.8% of voxelotor related material in plasma. Five further metabolites accounted for a total of 23% of voxelotor related material in plasma, with individual contributions up to 9%. All other metabolites were less than 5%.
Elimination
The major route of elimination of voxelotor is by metabolism with subsequent excretion of metabolites into urine and faeces. The excretion of unchanged voxelotor is minimal (< 1% of dose in urine). The geometric mean (%CV) terminal elimination half-life of voxelotor in patients with SCD is 38.7 hours (30.2%) with concentrations in plasma and whole blood declining in parallel. The apparent oral clearance of voxelotor was estimated as 6.1 L/h in plasma in patients with SCD.
Special populations
Patients with renal impairment
There was no clinically significant effect of renal function on the excretion of voxelotor in subjects without SCD and patients with SCD. Following a single 900 mg dose of voxelotor, whole blood exposures in subjects with severe renal impairment (eGFR < 30 mL/min/1.73 m2) were 25% lower compared to healthy controls. The unbound plasma concentrations were comparable. In patients with SCD, a trend for higher voxelotor exposure was observed with lower Cystatin C levels. Higher levels of Cystatin C typically observed with renal impairment were not associated with higher voxelotor exposure.
Voxelotor has not been evaluated in patients with ESRD requiring dialysis.
Patients with hepatic impairment
In plasma, the Cmax was 1.2-fold higher in subjects with mild hepatic impairment (Child Pugh A), 1.5-fold higher in subjects with moderate hepatic impairment (Child Pugh B) and 1.4-fold higher in subjects with severe hepatic impairment (Child Pugh C), and the AUCinf was 1.1-fold higher in subjects with mild hepatic impairment, 1.2-fold higher in subjects with moderate hepatic impairment and 1.9-fold higher in subjects with severe hepatic impairment. In whole blood, increase in exposure was similar to that in plasma. No dose adjustment is warranted in subjects with mild to moderate hepatic impairment, but it is recommended to reduce the daily dose of voxelotor to 1,000 mg in subjects with severe hepatic impairment (see section 4.2). The plasma and whole blood Cmax values in patients with severe hepatic impairment after dose adjustment are expected to be similar to those in patients with normal hepatic function treated at the recommended dose of 1,500 mg daily. The plasma and whole blood AUC are expected to be ~25% higher in subjects with severe hepatic impairment after dose adjustment compared to those in patients with normal hepatic function treated at the recommended dose of 1,500 mg daily.
Effect of gender, race, and body weight
No clinically significant differences in the pharmacokinetics of voxelotor were observed based on gender, race, and body weight (28 to 135 kg).
Effect of age
No clinically significant differences in the pharmacokinetics of voxelotor were observed based on age (12 to 59 years).
Effect of haematocrit
The blood-to-plasma partitioning of voxelotor increases with increasing haematocrit. As haematocrit increased from 30.5% in SCD patients (median at 1,500 mg daily) to the maximum haematocrit measured at 1,500 mg daily (35.1%), the blood-to-plasma partitioning increased from 14.8 to 16.4 (11% increase).
Patients with HbSC genotype
Voxelotor steady state whole blood AUC and Cmax were 50% and 45% higher in HbSC genotype patients (n=11) compared to HbSS genotype (n=220) patients and voxelotor steady state plasma AUC and Cmax were 23% and 15% higher in HbSC genotype patients compared to HbSS genotype patients.
In vitro drug interactions
CYP enzymes: In vitro voxelotor is an inhibitor and inducer of CYP2B6. In vitro, voxelotor is an inhibitor of CYP2C8, CYP2C9, CYP2C19 and CYP3A4. The clinical relevance is currently unknown (see section 4.5).
Transporter-mediated interactions: Voxelotor is not an inhibitor of P-gp, BCRP, OATP1B3, OCT2, OAT1, MATE2-K, or BSEP. Voxelotor act as an inhibitor of OATP1B1, OAT3 and MATE1 transporters (see section 4.5). Voxelotor is not a substrate of P-gp, BCRP, OATP1A2, OATP1B1, OATP1B3, or BSEP.
5.3 Preclinical safety data
Adverse reactions not observed in clinical studies, but seen in animals at exposure levels similar to clinical exposure levels and with possible relevance to clinical use were as follows:
Repeated dose toxicity
The major findings associated with repeat-dose administration of voxelotor was compensatory erythropoiesis, manifested as increased red blood cell mass (↑ RBC, HCT, Hb, RET) correlated microscopically with hypercellular bone marrow and splenic red pulp and increased splenic weight in rats, mice and cynomolgus monkeys. In monkeys, early stages of this effect were seen at dose levels comparable with clinical exposure (exposure multiple of ~0.6 based on the plasma Cmax values). Voxelotor also caused GI intolerance attributed to local irritation. Other findings attributed to voxelotor include induction of CYP enzymes in the liver of mice and rats, altered T cell-dependent antigen response in rodents and monkeys and prolongation of corrected QT (QTc) intervals in monkeys. Following the immunization with keyhole limpet hemocyanin (KLH), voxelotor caused the significantly reduced IgG (rats, monkeys) and IgM (monkeys) titres, a delayed peak in the antibody response (monkeys) and the changes in the relative lymphocyte distribution (rats). These effects were seen at the exposure multiple of the anticipated clinical exposure ~0.6 in monkeys and ~4.0 in rats based on plasma Cmax value. Treatment with voxelotor at the exposure multiple ~2.5 of the anticipated clinical exposure led to the QT and QTc intervals prolongation in monkeys.
Reproduction and development
Treatment of rats with voxelotor at exposure multiple ~4 of the anticipated clinical exposure caused a reduced sperm motility and an increased percentage of abnormal sperm, as well as an increased testicular and prostate weight and reduced seminal vesicles weight. These effects did not, however, affect the reproductive performance. Voxelotor was not teratogenic in rats and rabbits at exposure levels causing maternal toxicity (exposure multiple based on blood AUC of 2.8 in rats and 0.3 in rabbits). Voxelotor is excreted in milk of lactating rats. Milk exposure was up to 0.4-fold plasma exposure of the dams, leading to subsequent plasma exposure in pups. In the pre- and postnatal developmental toxicity study, adverse effects on the progeny, manifested as reduced pup viability index and persistently lower pup weight, were seen at the predicted exposure multiple of ~2.6 of the anticipated human exposure.
6. PHARMACEUTICAL PARTICULARS
6.1 List of excipients
Tablet core
Microcrystalline cellulose (E460) Croscarmellose sodium (E468) Sodium laurilsulfate (E487) Silica, colloidal anhydrous (E551) Magnesium stearate (E470b)
Tablet film-coating
Polyvinyl alcohol (E1203) Titanium dioxide (E171) Polyethylene glycol (E1521) Talc (E553b) Iron oxide yellow (E172)
6.2 Incompatibilities
Not applicable.
6.3 Shelf life
3 years.
6.4 Special precautions for storage
Store at or below 30° C.
6.5 Nature and contents of container
High-density polyethylene (HDPE) bottle with a polypropylene child-resistant cap and an aluminium induction seal. The bottle also contains a silica gel desiccant canister and polyester coil.
Pack-size of 90 film-coated tablets.
6.6 Special precautions for disposal and other handling
Any unused medicinal product or waste material should be disposed of in accordance with local requirements.
7. SCIENTIFIC OPINION HOLDER
Global Blood Therapeutic UK Limited
c/o Legalinx Ltd
Tallis House, 2 Tallis St, Temple
London EC4Y 0AB
8. EAMS NUMBER
54981/0001
9. DATE OF SCIENTIFIC OPINION
25/01/2022
Additional information
Physicians can enrol patients onto the EAMS program by registering through the Inceptua portal at https://portal.inceptua.com/#/login. When a treating physician requests patient entry into the EAMS scheme they will receive an electronic physician pack. The pack will include information on reporting of adverse events (AEs) and all the necessary forms and contact details
For patients receiving voxelotor under EAMS no specific clinical monitoring is required
Contact information
For medical information please login to https://gbtmedinfo.com/ or use the following email EAMSinquiry@gbt.com
or contact
Inceptua: at access@inceptua.com or by registering with Inceptua at https://portal.inceptua.com/#/login.
-
Adverse reactions were NCI Grades 1 or 2 except for Grade 3 diarrhoea (n=1), nausea (n=1), rash (n=1), rash generalized (n=3) and hypersensitivity (n=1). ↩
-
Abdominal pain included abdominal pain, abdominal pain upper, and abdominal pain lower. ↩
-
Rash included rash, urticaria, rash generalized, rash macular, rash maculo papular, rash pruritic, and rash papular. ↩